棍其勒-13味丸质量标准的建立
2021-09-20王秋桐崔圆圆王玉华
王秋桐,崔圆圆,王玉华
(1.内蒙古医科大学附属医院药物临床试验机构,内蒙古 呼和浩特010050;2.内蒙古医科大学药学院)
棍其勒-13味丸由诃子、广枣、红花、石膏、石榴、木香、枫香脂、胡黄连、丁香、豆蔻、肉豆蔻、炒马钱子、沉香十三味药组成的复方制剂。临床功效为镇赫依,清血热;用于胸部赫依、血相搏,“赫依”性心肺刺痛,胸闷、气短等症。棍其勒-13丸作为蒙医医院常用的蒙医经验方,目前没有相应的质量标准对其进行质量控制。为了更好的控制该制剂的质量,本实验对棍其勒-13味丸进行了定性鉴别和定量测定研究。
红花具有活血通经,散瘀止痛的功效[1]。参阅近几年中药红花的研究文献,其主要的化学成分是黄酮类、甾体、酚酸类、双醇、木脂素、查尔酮、炔类以及挥发油等[2,3]。查尔酮类成分红花黄色素是红花现在已知的主要活性成分,根据官能团的不同,化学结构的分为红花黄色素A和红花黄色素B两种成分[2],羟基红花黄色素A(hydroxysafflor yellow A,HSYA)是红花中最具药理活性的水溶性成分,多年来一直是科研的热点,临床研究表明,其具有抗凝血、抗氧化、预防脑缺血、保护血管内皮细胞和神经等作用,是2020版中国药典收录红花药材的质量控制指标成分之一[4,5]。故选择HSYA作为指标成分,采用HPLC法对处方中红花的指标性成分HSYA进行含量测定。
1 仪器与试药
1.1 仪器

仪器高效液相色谱仪电子天平电子天平超声波清洗器超纯水系统多功能粉碎机薄层色谱成像仪电脑UIS生物显微镜型号Waters e2695 Mettler-Toledo MS105DU Mettler-Toledo XPR10 SBL-22DT Heal Force NW15UV FW400A GoodSee-10 XSP-BM-30AD厂家沃特世科技有限公司梅特勒-托利多仪器有限公司梅特勒-托利多仪器有限公司宁波新芝生物科技股份有限公司河南省予华仪器有限公司北京材茂科技有限公司上海科哲生化科技有限公司上海彼爱姆光学仪器制造有限公司
1.2 试剂与试药
对照品:HSYA(111838-201403),去氢木香内酯(111525-201912)。
对照药材:枫香脂(121637-201201),均购于中检院。
供试品:棍其勒-13味丸(201908001、201908002、201908003),由呼伦贝尔市蒙医医院生产;自制样品(20190801)。
试剂:甲醇、乙腈、三乙胺为色谱纯,其他为分析纯。
2 方法与结果
2.1 显微鉴别
取本品,置显微镜下观察:石细胞无色,椭圆形,壁厚,孔沟细密(石榴)。脂肪油滴众多,加水合氯醛试液加热后渐形成针簇状结晶(肉豆蔻)。菊糖团块形状不规则,有时可见微细放射状纹理(木香)。橄榄形、椭圆形或类圆形的花粉粒,具3个萌发孔,直径约至60μm,外壁有齿状突起(红花)。石细胞成群,呈长卵形、类圆形、长条形或长方形,孔沟细密而明显(诃子)。果皮表皮细胞成片,表面观类多角形或类圆形,胞腔内含颗粒状物(广枣)。不规则片状结晶,无色,有平直纹理(石膏)。具缘纹孔导管,纹孔密,内含淡黄色或黄棕色树脂状物(沉香)。显微特征照片见图1。
2.2 薄层色谱鉴别
2.2.1 枫香脂薄层色谱鉴别
取本品2.0g,粉碎,加10mL甲醇,用超声处理20分钟,取上清液作为供试品溶液。另取枫香脂对照药材0.2g和阴性供试品2.0g,同法分别制成对照药材溶液和阴性对照溶液。点样量各5μL,薄层板为硅胶GF254薄层板,展开剂为正己烷-乙酸乙酯-石油醚(60~90℃)-冰醋酸(6:3:2:0.2),检视方式为在紫外灯(254nm)下检视。薄层色谱图见图2。
图2的结果显示,枫香脂对照药材溶液显示1个主斑点,供试品溶液在与其对应位

图2 枫香脂TLC鉴别图Tab.2 Liquidambar Formosana Hance TLC Identification Chart
置处显相同颜色的斑点;阴性对照对测定没有干扰。
2.2.2 木香薄层色谱鉴别
取本品2.5g,粉碎,加10mL甲醇,用超声处理0.5小时,过滤,作为供试品溶液。另取去氢木香内酯对照品,加甲醇制成0.5mg/mL的对照品溶液。取阴性供试品2.5g,同法制成阴性对照溶液。点样量各5μL,薄层板为硅胶G薄层板,展开剂为环己烷-甲酸乙酯-甲酸(15:5:1),检视方式为喷1%香草醛硫酸溶液,加热至斑点显色清晰。薄层色谱图见图3。
图3的结果显示,去氢木香内酯对照品溶液显示1个斑点,供试品溶液在与对照品溶液Rf值相同的位置上显相同颜色的斑点;阴性对照对测定没有干扰。
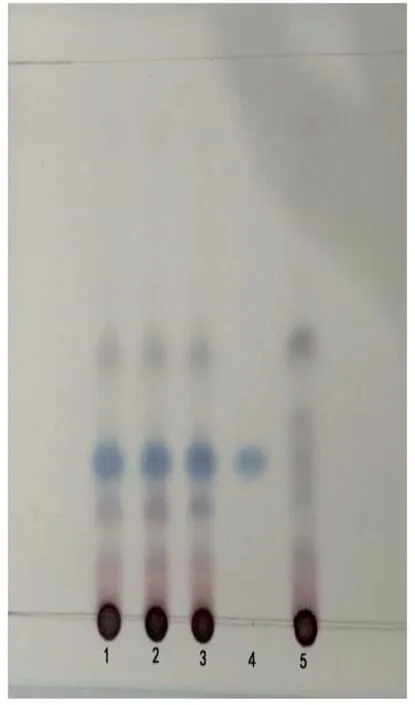
图3 木香TLC鉴别图Tab.3 Aucklandia lappa Decne TLC Identification Chart
2.3 含量测定
2.3.1 色谱条件
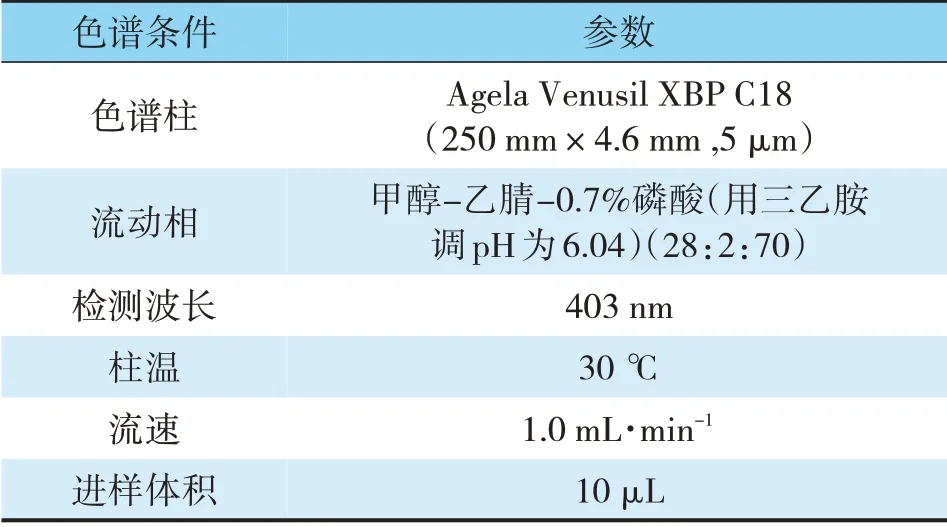
色谱条件色谱柱流动相检测波长柱温流速进样体积参数Agela Venusil XBP C18(250 mm×4.6 mm,5 μm)甲醇-乙腈-0.7%磷酸(用三乙胺调pH为6.04)(28:2:70)403 nm 30℃1.0 mL·min-1 10 μL
2.3.2 溶液的制备
将本品粉碎,精密称取1.7g,精密加25%甲醇25ml,称重,超声40分钟,放冷,再称重,用25%甲醇补,离心5分钟(转速7000r/min),取上清液作为供试品溶液。再取HSYA对照品,加25%甲醇制成56.79μg/mL的对照品溶液。取阴性供试品1.7g,同法制成阴性对照溶液。
2.3.3 方法学考察
2.3.3.1 专属性实验
在“2.3.1”项色谱条件下将上述制备的三种溶液进液相测定,记录色谱图。色谱图见图4。
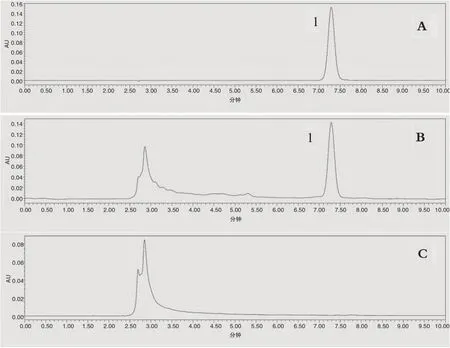
图4 专属性试验色谱图Tab.4 Exclusive experiment chromatogram
图4结果显示,在相同的保留时间处,供试品和对照品色谱图中均有色谱峰出现;阴性对照色谱图无色谱峰出现,专属性强。
2.3.3.2 线性关系考察
取上述制备的对照品溶液,分别精密吸取2μL、5μL、10μL、15μL、20μL和25μL进液相测定,以峰面积对进样量进行分析,结果见表1和图5。
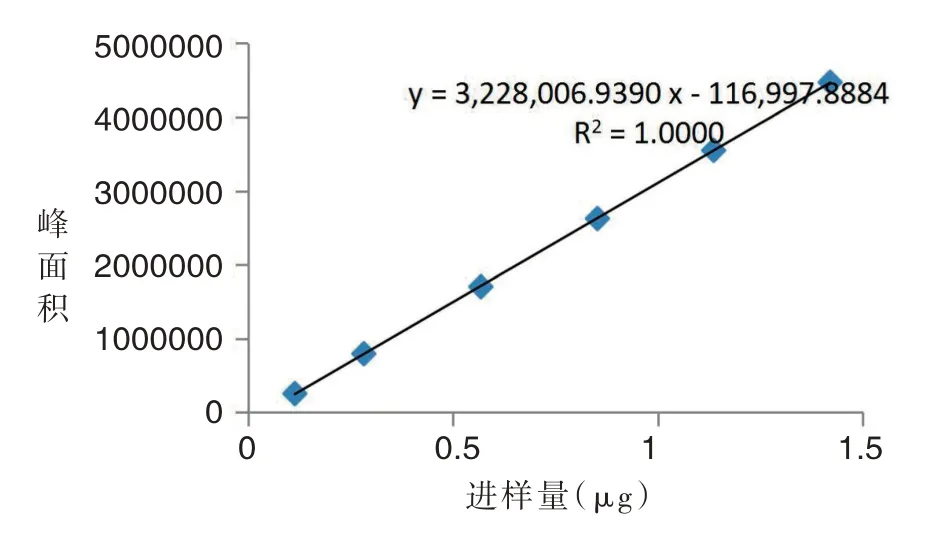
图5 标准曲线图Tab.5 Standard curve chart
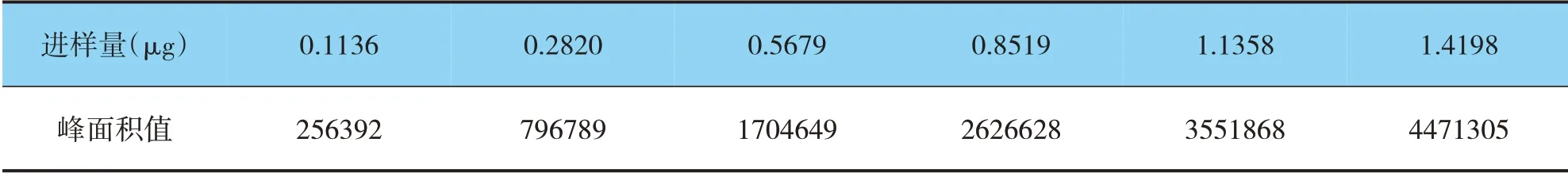
表1 标准曲线数值表Fig.1 Standard curve value table
结果线性关系较好,回归方程y=3228006.9390x-116997.8884(r=1.0000)。
2.3.3.3 精密度试验
取一份供试品溶液连续进样6针进行测定,结果HSYA峰面积值的RSD为0.63%。仪器精密度好。
2.3.3.4 稳定性试验
取一份供试品溶液,分别在溶液制备后的0、2、4、8、10和12小时进样测定,结果HSYA峰面积值RSD为1.27%。表明12小时内供试品溶液稳定。
2.3.3.5 重复性试验
取同一批号(批号201908001)供试品6份,制成供试品溶液后进样分析,计算含量。结果HSYA的平均含量为0.87mg·g-1,含量RSD值为0.29%。该方法的重复性好。
2.5.6 加样回收试验
取供试品(批号201908001,含量为0.87 mg·g-1)9份,精密称取0.85g,分成三组,每组三份,每组分别精密加入约相当于供试品含有量的50%、100%、150%的HSYA对照品及25%甲醇25mL,再按上述方法制备供试品溶液,进样分析,计算含量并计算回收率。结果见表2。
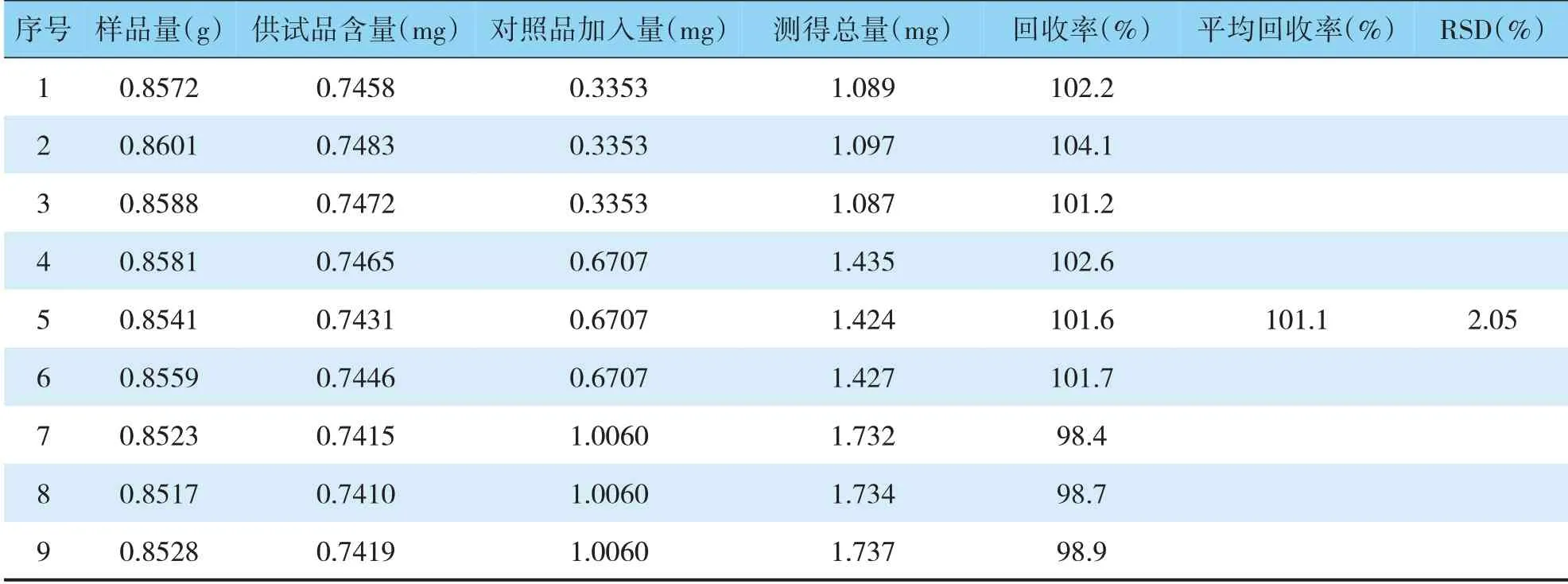
表2 加样回收试验结果表Fig.2 Test results of Sample recovery test
表2结果显示,HSYA的平均回收率为101.1%,RSD为2.05%,表明该方法准确、可靠。
2.5.7 耐用性试验
取供试品两份,按上述拟定的方法制备供试品溶液,用不同厂家色谱柱和柱温箱子不同温度下进行测定。结果在使用不同厂家的色谱柱测定时,HSYA色谱峰分离度均良好,且其含量测定结果之间没有显著差异。在柱温箱温度不同(25℃、30℃、35℃)的情况下对实验结果的影响较小,耐用性好。
2.5.8 样品含量测定
取3批样品和1份自制样品(批号201908001,201908002,201908003,201908010)各2份,制备供试品溶液后进样测定,计算含量。结果见表3。

表3 含量测定结果表Fig.3 Assay result
表3的结果显示,3批棍其勒-13味丸样品中HSYA含量很接近,自制样品含量稍高些。
3 讨论
3.1 流动相的调整
参照《中国药典》2020年版一部“红花”项下的测定方法[1],选择甲醇-0.7%磷酸-乙腈(26:72:2)为流动相进行实验,但HSYA峰型不对称且拖尾严重。加三乙胺调节0.7%磷酸溶液pH值至6.0后,供试品色谱图中的HSYA峰的对称性由拖尾变成前沿峰,故调整流动相比例为甲醇-乙腈-0.7%磷酸溶液(28:2:70)。此时HSYA色谱峰的拖尾因子在0.95-1.05之间且与相邻峰达到较好分离,理论板数较高,并具有适宜的保留时间。经以上试验确定流动相为甲醇-乙腈0.7%磷酸水溶液(28:2:70)[6~8]。流动相中的0.7%磷酸水溶液的pH值为6(用三乙胺调节)。
3.2 样品粉碎粒度的选择
以《中国药典》2020年版一部“红花”含测项下的粉碎细度为参考依据,确定本品粉碎细度为研细后过三号筛。
3.3 溶剂用量的确定
《中国药典》2020年版一部“红花”含测项下的红花称样量为0.4g,加提取溶剂50mL。根据棍其勒-13味丸处方总量和处方中红花的量计算,称取棍其勒-13味丸1.7g含红花药材0.2g,故选择加提取溶剂25mL。
3.4 超声提取时间的选择
为确定超声提取时间对提取效率的影响,本试验研究比较了提取时间为30 min、40 min和50 min时的提取效率。结果超声提取30 min时含量偏低,超声提取40 min和50 min时,含量基本相同。故确定超声时间为40 min。
