三味板蓝根颗粒的质量标准研究Δ
2021-09-16梁国成陈斯宁陈舒茵杨红梅黄小鸥覃忠桂韦世民
梁国成,陈斯宁,陈舒茵,杨红梅,黄小鸥,覃忠桂,3,秦 辛,韦世民,3
(1.广西中医药大学附属瑞康医院药物研发中心,广西 南宁 530011; 2.广西大学化学化工学院,广西 南宁 530004; 3.广西壮瑶药工程技术研究中心,广西 南宁 530200)
三味板蓝根颗粒是广西中医药大学附属瑞康医院临床常用的医院制剂(批准文号:桂药制字Z01060082),其处方简单,仅由板蓝根、贯众和山芝麻3味中药组成,其中板蓝根为传统的清热解毒药,贯众与山芝麻是广西壮族自治区特色壮瑶药,该制剂极具地方特色,具有清热解毒、凉血利咽和消肿的功效,在临床常被用于扁桃体炎、腮腺炎、咽喉肿痛以及病毒性肝炎等的防治。三味板蓝根颗粒在临床上已有超过四十年的使用历史,其原有的质量标准偏低,已无法适应如今中药、民族药制剂发展以及中药制剂现代化的相关要求。因此,本研究开展了医院制剂三味板蓝根颗粒的质量标准提升研究,在前期工艺改良的前提下,进一步增加薄层色谱法(thin layer chromatography,TLC)、紫外-可见分光光度法(ultraviolet and visible spectrophotometry,UV-Vis)和高效液相色谱法(high performance liquid chromatography,HPLC)等检测技术和手段,探讨提升原制剂的质量标准,为控制该制剂的质量提供参考和依据。
1 材料
1.1 仪器
L6型紫外可见分光计[上海仪电(集团)有限公司];D27型超声波清洗机(广东固特超声股份有限公司);JJ500型电子天平(常熟市双杰测试仪器厂);AE240型分析天平[梅特勒-托利多仪器(上海)有限公司];DK-S26型电热恒温水浴锅(上海精宏实验设备有限公司);101-38型数显式电热恒温干燥箱(上海沪越实验仪器有限公司);LC-20A型高效液相色谱仪(日本岛津公司)。
1.2 药品与试剂
三味板蓝根颗粒处方药材均购自广西柳州百草堂中药饮片厂有限责任公司(板蓝根批号为20200209,贯众批号为20170902,山芝麻批号为20170814);三味板蓝根颗粒(自制,批号为20200511-TS01、20200511-TS02、20200511-TS03、20200511-TS04、20200511-TS05和20200511-TS06);(R,S)-告依春对照品(中国食品药品检定研究院,批号为111753-201706);β-谷甾醇(中国食品药品检定研究院,批号为110851-201909);靛玉红(中国食品药品检定研究院,批号为110717-201805)。石油醚(60~90 ℃)(成都市科隆化学品有限公司,批号为2018101701);乙酸乙酯(成都市科隆化学品有限公司,批号为2018120501);甲醇(成都市科隆化学品有限公司,批号为2020041402);苯酚(成都金山化学试剂有限公司,批号为20170816);冰醋酸(上海吴泾化工有限公司,批号为20110101);氢氧化钠(天津市大茂化学试剂厂,批号为20191101);碳酸氢钠(新兴凌云医药化工有限公司,批号为03832550),均为分析纯。无水乙醇(成都市科隆化学品有限公司,批号为2020082702);色谱甲醇(美国Tedia公司,批号为14060185);娃哈哈纯净水(杭州娃哈哈集团,批号为202008171113GS 07645);10%硫酸乙醇溶液(自制);纯化水(自制)。
2 方法与结果
2.1 TLC鉴别研究与结果
2.1.1 板蓝根的TLC鉴别:称取三味板蓝根颗粒成品颗粒10 g,加入80%甲醇50 ml溶解并超声处理30 min(频率为40 kHz,功率为600 W),过滤,将所得滤液蒸干,残渣加甲醇1 ml溶解,作为供试品溶液。另取板蓝根对照药材2 g和缺板蓝根的阴性样品颗粒10 g,分别加入80%甲醇10和50 ml,同法制成板蓝根对照药材溶液和阴性样品溶液。再精密称取(R,S)-告依春对照品,加入甲醇溶解制成0.5 mg/ml的溶液,作为对照品溶液。按照《中华人民共和国药典:一部》(2020年版)[1]中“板蓝根”鉴别项、《中华人民共和国药典:四部》(2020年版)[2]通则0502中TLC法进行试验,吸取上述4种溶液各5 μl,分别点于同一硅胶GF254薄层板上,以石油醚(60~90 ℃)-乙酸乙酯(V∶V=1∶1)为展开剂,在层析缸中预饱和后展开,取出晾干后置于紫外光灯(254 nm)下检视。供试品色谱中,在与对照药材及对照品色谱相应位置上,分别显相同颜色斑点,且阴性无干扰,见图1。
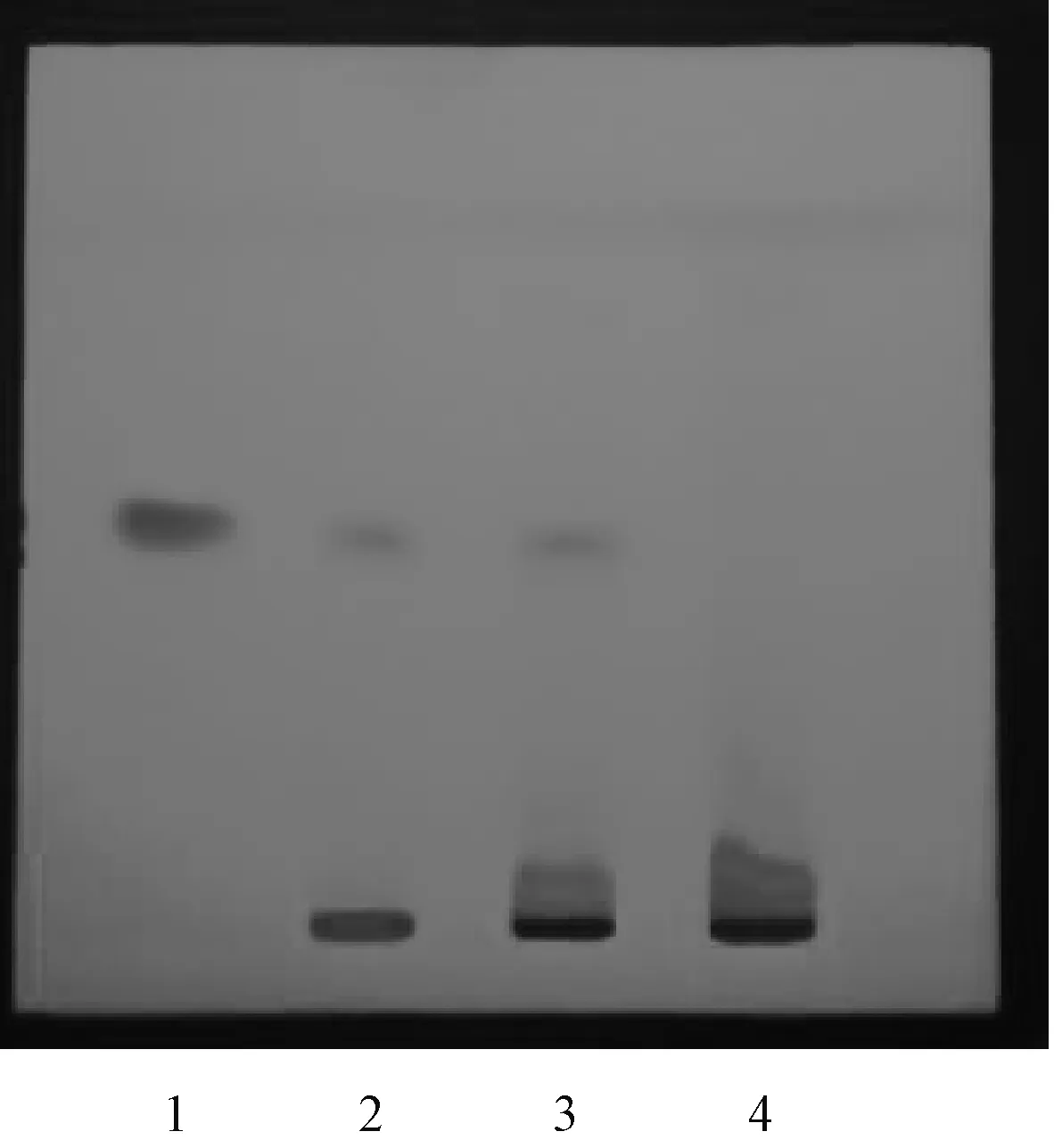
1.(R,S)-告依春对照品;2.板蓝根对照药材;3.三味板蓝根颗粒供试品;4.阴性样品1.(R,S)-goitrin reference; 2.radix isatidis reference; 3.test sample of Sanwei radix isatidis granules; 4.negative sample图1 板蓝根的TLC色谱鉴别图Fig 1 TLC chromatographic identification of radix isatidis
2.1.2 山芝麻的TLC鉴别:称取三味板蓝根颗粒成品颗粒10 g,加入甲醇50 ml溶解并超声处理30 min(频率为40 kHz,功率为600 W),放冷,过滤,将所得滤液蒸干,残渣加入甲醇1 ml溶解,作为供试品溶液。另取山芝麻对照药材2 g和缺山芝麻的阴性样品颗粒10 g,分别加入甲醇10和50 ml,同法制成对照药材溶液和阴性样品溶液。再精密称取β-谷甾醇对照品,加入甲醇溶解制成0.5 mg/ml的溶液,作为对照品溶液。按照《中华人民共和国药典:四部》(2020年版)通则0502中TLC法进行试验,吸取上述4种溶液各5 μl,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-乙酸乙酯(V∶V=7∶1)为展开剂,在层析缸中预饱和后展开,取出晾干后均匀喷以10%硫酸乙醇溶液,放置于105 ℃烘箱中加热至斑点显色清晰[3-4]。供试品色谱中,在与对照药材及对照品色谱相应位置上,分别显相同颜色斑点,且阴性无干扰,见图2。

1.β-谷甾醇对照品;2.三味板蓝根颗粒供试品;3.山芝麻对照药材;4.阴性样品1.β-sitosterol reference; 2.test sample of Sanwei radix isatidis granules; 3.screwtree root reference; 4. negative sample图2 贯众的TLC色谱鉴别图Fig 2 TLC chromatographic identification of cyrtomium fortunei
2.2 多糖含量测定研究与结果
2.2.1 溶液的制备:(1)对照品溶液的制备。取适量先于105 ℃下干燥至恒重的葡萄糖对照品,精密称取25 mg置于25 ml容量瓶中,加水稀释至刻度,摇匀,再取5 ml溶液转移至25 ml容量瓶中并加水稀释至刻度,摇匀,制成质量浓度为0.2 mg/ml的对照品溶液。(2)供试品溶液的制备。称取三味板蓝根颗粒10 g,加入1 ml/mol的氢氧化钠溶液50 ml,在80 ℃的水中超声处理50 min(频率为40 kHz,功率为600 W),用10%醋酸溶液调节pH至中性,过滤,精密吸取续滤液1 ml转移至50 ml容量瓶中并加水稀释至刻度,摇匀,作为供试品溶液。(3)5%苯酚溶液的制备。称取苯酚100 g,加铝粉0.1 g及碳酸氢钠粉末0.05 g,采用常压蒸馏法收集181~182 ℃的馏分,称取该冷凝后的馏分5 g,加水定容于100 ml的棕色容量瓶中,即得5%苯酚溶液。
2.2.2 检测波长的确定:按照《中华人民共和国药典:四部》(2020年版)通则0401中UV-Vis法进行试验[2]。精密吸取对照品溶液和供试品溶液各0.5 ml,分别置于10 ml容量瓶中,分别加入5%苯酚溶液1 ml,摇匀,再分别加入浓硫酸5 ml边加边震荡,移至45 ℃水浴中放置30 min后再移至冰水浴中放置30 min。放置至室温(25 ℃)后,在紫外-可见分光光度计上测定波长400~500 nm之间的吸光度,结果显示,两者均在486 nm处有最大吸收(见图3—4),故选择486 nm为测定波长。

图3 对照品溶液不同波长的吸光度曲线Fig 3 Absorbance curves of reference solution at different wavelengths

图4 供试品溶液不同波长的吸光度曲线Fig 4 Absorbance curves of test solution at different wavelengths
2.2.3 方法学验证:(1)线性关系的确定。精密吸取葡萄糖对照品溶液0.1、0.2、0.3、0.4和0.5 ml,分别置于10 ml容量瓶中,补加蒸馏水至1.0 ml,加入5%苯酚溶液1 ml,摇匀,再加入浓硫酸5 ml,再次混合均匀后,转移至45 ℃水浴中保温30 min,取出冷却至室温,于486 nm处测定吸光度。以对照品质量浓度为横坐标(X),以吸光度值为纵坐标(Y),绘制标准曲线,得到回归方程为Y=6.215X+0.059 3,R2=0.999 1。结果表明,葡萄糖对照品溶液在0.02~0.10 mg/ml范围内线性关系良好。(2)精密度试验。取葡萄糖对照品,按照“2.2.1”项下对照品溶液的制备方法,制得葡萄糖对照品溶液,按照“2.2.2”项下方法处理后测定,连续测定吸光度6次。结果显示,葡萄糖对照品溶液的吸光度分别为0.722、0.714、0.718、0.718、0.717和0.717,RSD为0.33%,表明仪器精密度良好。(3)重复性试验。称取6份同一批三味板蓝根颗粒(批号为20200511-TS01)适量,按照“2.2.1”项下供试品溶液的制备方法,制得6份供试品溶液,按照“2.2.2”项下方法处理后测定,记录各吸光度。结果显示,各样品的吸光度分别为0.450、0.434、0.450、0.439、0.468和0.443,RSD为2.43%,表明方法重复性良好。(4)稳定性试验。取同一批三味板蓝根颗粒(批号为20200511-TS01)供试品溶液,于室温下放置,分别于0、30、60、90、120和150 min时按照“2.2.2”项下拟定条件测定,记录吸光度。结果显示,各样品的吸光度分别为0.450、0.444、0.443、0.444、0.440和0.428,RSD为1.52%,表明三味板蓝根颗粒供试品溶液试样在室温下放置150 min内的稳定性良好。(5)回收率试验。取已知含量的同一批三味板蓝根颗粒(批号为20200511-TS01)9份,每份10 g,分别按低剂量、中剂量和高剂量加入葡萄糖对照品0.225 0、0.450 0和0.900 0 mg各3份,使每3份葡萄糖对照品成分的含量约为样品颗粒葡萄糖含有量的0.5、1.0和1.5倍,按照“2.2.1”项下供试品溶液的制备方法,制得样品溶液,再按“2.2.2”项下拟定条件测定,记录吸光度。结果显示,低剂量、中剂量和高剂量3种加入剂量的样品溶液的平均加样回收率分别为101.54%、101.48%和101.54%,RSD分别为0.70%、0.71%和0.45%,表明方法准确性良好。
2.2.4 多糖含量测定:取6批三味板蓝根颗粒样品,按照“2.2.1”项下供试品溶液的制备方法,制得样品溶液,再按照“2.2.2”项下拟定条件测定,重复进样3次,记录吸光度。结果显示,6批三味板蓝根颗粒中多糖含量分别为6.31%、6.27%、6.25%、6.33%、6.37%和6.35%,平均含量为6.31%,RSD为0.67%。
2.3 (R,S)-告依春含量测定研究与结果
2.3.1 溶液的制备:(1)对照品溶液的制备。精密称取(R,S)-告依春对照品2.0 mg,置于50 ml容量瓶中,以甲醇溶解并定容至刻度,再精密吸取20 ml,置于50 ml容量瓶中,以甲醇定容至刻度,摇匀,制成质量浓度为16 μg/ml的溶液,以0.45 μm微孔滤膜滤过,取续滤液作为对照品溶液。(2)供试品溶液的制备。取三味板蓝根颗粒样品颗粒5 g,精密称定,置于具塞三角锥形瓶中,加纯化水25 ml,精密称定质量,超声处理30 min(频率为40 kHz,功率为600 W),放冷至室温,再称定质量,以纯化水补足减失的质量,摇匀,取上清液以0.45 μm微孔滤膜滤过,取续滤液作为供试品溶液。(3)阴性样品溶液的制备。取缺板蓝根药材的阴性样品颗粒5 g,按照“2.3.1”项下供试品溶液的制备方法,制得阴性样品溶液。
2.3.2 色谱条件的确定:色谱柱为Waters Symmetry Shield RP18柱(250 mm×4.6 mm,5 μm),流动相为甲醇-水(V∶V=15∶85),体积流量为1.0 ml/min,柱温为25 ℃,检测波长为245 nm,进样量为5 μl。按照《中华人民共和国药典:四部》(2020年版)[2]通则0512中HPLC法进行试验。HPLC图见图5—7。

图5 (R,S)-告依春对照品的HPLC色谱图Fig 5 HPLC chromatogram of (R,S)-goitrin reference
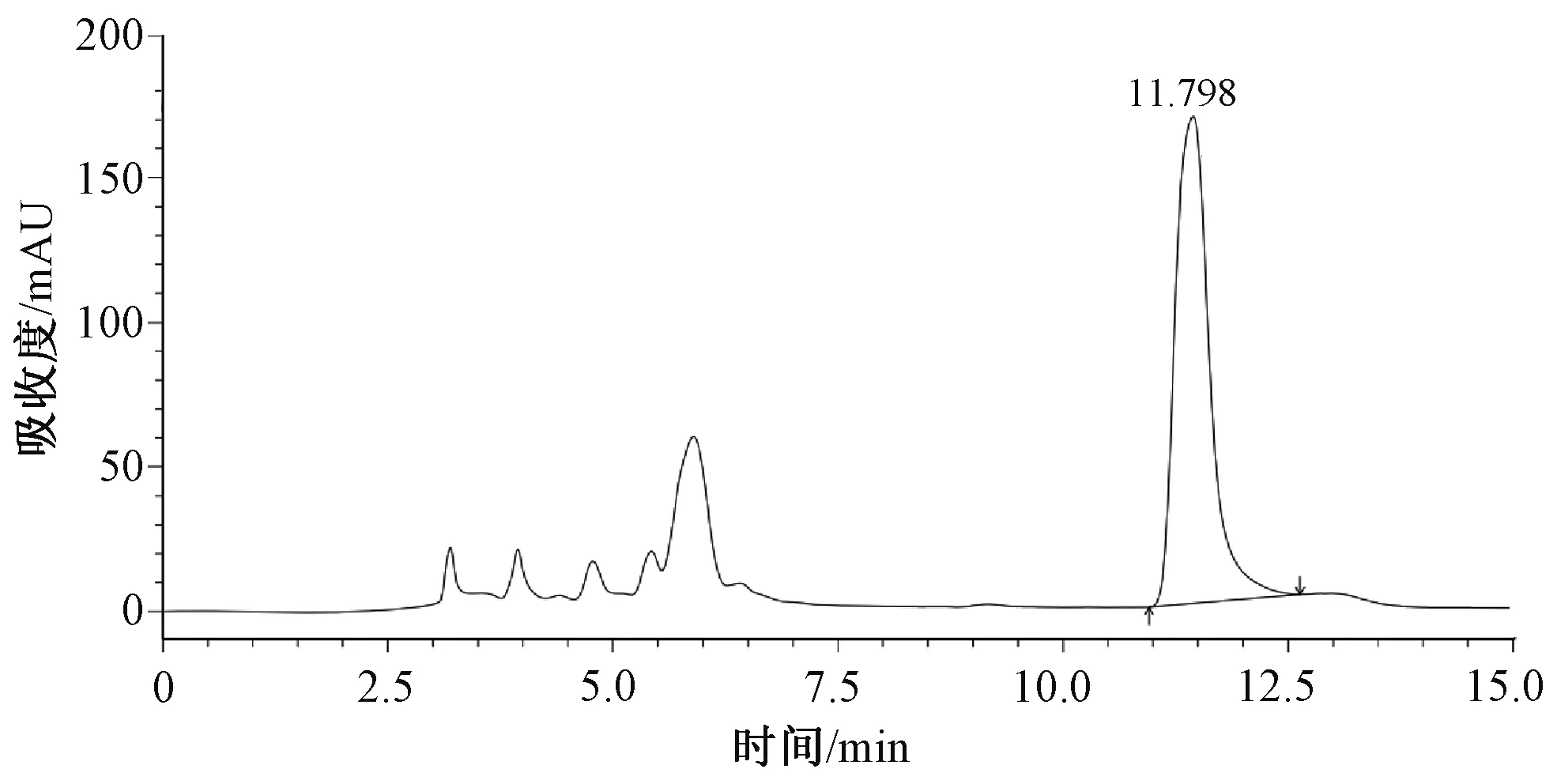
图6 供试品的HPLC色谱图Fig 6 HPLC chromatogram of test sample

图7 阴性样品的HPLC色谱图Fig 7 HPLC chromatogram of negative sample
2.3.3 方法学验证:(1)线性及线性关系。精密吸取“2.3.1”项下制备的(R,S)-告依春对照品溶液1.0、2.0、3.0、4.0和5.0 ml,分别置于10 ml容量瓶中,以甲醇稀释定容至刻度,摇匀,经0.45 μm微孔滤膜滤过,按照“2.3.2”项下拟定的色谱条件进样测定,平行测定3次,记录色谱图。以峰面积积分值(Y)为纵坐标,以进样量(X,μg)为横坐标,进行线性回归,得回归方程Y=6.0×106X+155 05,R2=0.999 7。结果表明,在0.008~0.045 μg内线性关系良好。(2)精密度试验。取“2.3.1”项下制备的(R,S)-告依春对照品溶液,按照“2.3.2”项下拟定的色谱条件连续进样6次,记录各色谱峰面积。结果显示,色谱峰面积的RSD为1.81%,表明仪器精密度良好。(3)重复性试验。分别称取6份同一批(批号为20200511-TS01)三味板蓝根颗粒处方药材适量,按照“2.3.1”项下供试品溶液的制备方法,制备6份供试品溶液,按照“2.3.2”项下拟定的色谱条件进行测定,记录色谱峰面积并计算(R,S)-告依春含量。结果显示,(R,S)-告依春平均含量为0.036 9 mg/g,RSD为1.46%,表明方法重复性良好。(4)稳定性试验。取同一批“2.3.1”项下制备的三味板蓝根颗粒(批号为20200511-TS01)供试品溶液,于室温下放置,分别于0、1、2、4、8、12和24 h,按“2.3.2”项下拟定的色谱条件进行测定,记录色谱峰面积。结果显示,RSD为0.51%,表明三味板蓝根颗粒供试品溶液试样在室温下放置24 h内的稳定性良好。(5)回收率试验。取已知含量的同一批三味板蓝根颗粒(批号为20200511-TS01)9份,每份5 g,分别按照低剂量、中剂量和高剂量加入(R,S)-告依春对照品0.092 3、0.184 5和0.276 8 mg各3份,使每3份(R,S)-告依春成分的含量约为样品颗粒(R,S)-告依春含有量的0.5、1.0和1.5倍,按照“2.3.1”项下供试品溶液的制备方法,制得样品溶液,再按照“2.3.2”项下拟定的色谱条件测定,记录色谱峰面积。结果显示,低剂量、中剂量和高剂量3种加入剂量的样品溶液的平均加样回收率分别为98.81%、100.16%和101.06%,RSD分别为1.32%、0.84%和0.80%,表明方法准确性良好。
2.3.4 (R,S)-告依春含量测定:取6批三味板蓝根颗粒样品,按照“2.3.1”项下供试品溶液的制备方法,制得样品溶液,再按照“2.3.2”项下拟定的色谱条件进行测定,重复进样3次,记录色谱峰面积。结果显示,6批三味板蓝根颗粒中(R,S)-告依春的含量分别为0.034 6、0.037 2、0.038 0、0.035 9、0.031 4和0.035 7 mg/g,平均含量为0.035 5 mg/g,RSD为5.98%。
3 讨论
三味板蓝根颗粒为广西中医药大学附属瑞康医院临床传统的中药复方制剂,处方组成虽然简单,但是四十余年的临床应用证实其药效确切,且该制剂为颗粒剂,携带、服用方便。该制剂处方中含广西地区特色中药贯众、特色壮瑶药山芝麻,极具民族地方特色。因此,加强对该制剂的深入研究与二次开发,以及质量标准研究和提升,对深入挖掘地方中药和民族药,推广、壮大广西地区特色中药和民族药具有积极的作用。
本研究中,对三味板蓝根颗粒处方药材进行了TLC定性鉴别。在板蓝根的定性鉴别中,同时考察了3、5和10 μl的点样量,发现点样量为3 μl时斑点显色较淡,点样量为5和10 μl时斑点显色清晰,故点样量定为5 μl即可达到要求。在山芝麻的定性鉴别色谱研究中,对供试品的制备进行了比较,当以水、甲醇和正丁醇等为溶剂进行供试品制备时,得到的供试品杂质较多,造成拖尾,尤其以甲醇为溶剂进行提取时杂质最多,与相关文献报道相符[5]。以乙酸乙酯进行供试品溶液的制备时,TLC色谱图中斑点清晰。结合相关文献,山芝麻的乙酸乙酯部位杂质干扰较少,HPLC色谱图显示各成分色谱峰分离度良好[6];相关抑菌试验结果表明,山芝麻的乙酸乙酯部位为抑菌活性部位之一[7]。因此,本研究选择乙酸乙酯进行供试品溶液的制备。此外,尝试对处方中贯众药材进行了TLC定性鉴别,采用环己烷法制备供试品溶液,以正己烷-三氯甲烷-甲醇(V∶V∶V=30∶15∶1)展开,以0.3%坚劳蓝BB盐的稀乙醇溶液显色,效果不理想,斑点不清晰,干扰多,故暂不列入本次质量标准范围,留待后期持续研究加以完善。
文献资料显示,板蓝根(Isatidis Radix)为十字花科植物菘蓝IsatisindigoticaFort. 的干燥根[1];主要含多糖类、生物碱类和黄酮类等多种活性成分,多糖为其主要有效成分之一,具有抗感染、抗肿瘤和免疫调节等多种生物活性[8-10]。《广西中药材标准》(1990年版)[11]收录了贯众(Rhizoma Osmundae Seu Blechnj Braineae),为紫萁蕨科华南紫萁OsmundavachelliiHook.、乌毛蕨科植物乌毛蕨BlechnumorientaleL. 或苏铁蕨Braineainsignis(Hook.) J. Sm. 的干燥根状茎;主要含多糖类、黄酮类和生物碱类等成分[12-13]。山芝麻(Radix seu herba Helicteris)为梧桐科植物山芝麻HelicteresangustifoliaL. 的干燥根或全株[14];主要含多糖类、三萜类和香豆素等成分[15-16]。现代药理研究结果证实,植物多糖具有多种药理活性,如增强免疫、抗氧化和降血糖等[17-19]。故本研究质量标准中,以多糖含量作为总物质含量测定指标之一。此外,关于多糖显色测定方法,苯酚-硫酸法为测定多糖含量较经典、有效的方法之一。本研究中使用氢氧化钠提取多糖,为避免多糖在碱性溶液中不稳定水解,加入10%醋酸调节pH至中性。通过全波长扫描选定了486 nm为测定波长,与相关文献中多糖的检测波长存在稍微差异[20-21],可能与试验时实际情况及环境相关。稳定性试验中发现,多糖显色后在15 min内稳定,故应在显色15 min内完成测定,避免误差。
综上所述,本研究为医院制剂三味板蓝根颗粒建立了较为完善的质量标准,且检验方法准确、重复性好、可操作性强,为今后三味板蓝根颗粒的质量标准提升及修订提供了依据。
