A simplified LC-MS/MS method for the quantification of the cardiovascular disease biomarker trimethylamine-N-oxide and its precursors
2021-09-14KatharinaRoxSilkeRathDietmarPieperMariusVitalMarkBronstrup
Katharina Rox , Silke Rath , Dietmar H. Pieper , Marius Vital ,d, Mark Br¨onstrup ,*
a Department of Chemical Biology (CBIO), Helmholtz Centre for Infection Research (HZI), 38124, Braunschweig, Germany
b German Center for Infection Research (DZIF), Partner Site Hannover-Braunschweig, 38124, Braunschweig, Germany
c Research Group Microbial Interactions and Processes (MINP), Helmholtz Centre for Infection Research (HZI), 38124, Braunschweig, Germany
d Institute for Medical Microbiology and Hospital Epidemiology, Hannover Medical School (MHH), 30625, Hannover, Germany
Keywords:TMAO Atherosclerosis Biomarker Carnitine Choline Betaine LC-MS/MS
ABSTRACT Trimethylamine-N-oxide (TMAO) has emerged as a potential biomarker for atherosclerosis and the development of cardiovascular diseases (CVDs). Although several clinical studies have shown striking associations of TMAO levels with atherosclerosis and CVDs,TMAO determinations are not clinical routine yet. The current methodology relies on isotope-labeled internal standards, which adds to pre-analytical complexity and costs for the quantification of TMAO and its precursors carnitine, betaine or choline.Here, we report a liquid chromatography-tandem mass spectrometry based method that is fast(throughput up to 240 samples/day),consumes low sample volumes(e.g.,from a finger prick),and does not require isotope-labeled standards.We circumvented the analytical problem posed by the presence of endogenous TMAO and its precursors in human plasma by using an artificial plasma matrix for calibration.We cross-validated the results obtained using an artificial matrix with those using mouse plasma matrix and demonstrated that TMAO, carnitine, betaine and choline were accurately quantified in ‘reallife’human plasma samples from healthy volunteers, obtained either from a finger prick or from venous puncture. Additionally, we assessed the stability of samples stored at-20 °C and room temperature.Whereas all metabolites were stable at-20 °C, increasing concentrations of choline were determined when stored at room temperature. Our method will facilitate the establishment of TMAO as a routine clinical biomarker in hematology in order to assess the risk for CVDs development,or to monitor disease progression and intervention effects.
1. Introduction
In recent years, trimethylamine-N-oxide (TMAO) has emerged as a potential biomarker for the development of cardiovascular diseases(CVDs)[1-4].Food rich in phosphatidylcholine such as red meat or eggs is the principal source for choline and other trimethylamine(TMA)-containing components,e.g.,betaine or carnitine[5,6].In the gut,choline,betaine,carnitine and similar components with a TMA-motif are catabolized to free TMA by intestinal microbes[2].TMA itself is then rapidly absorbed and metabolized by hepatic flavin monooxygenase 3 to TMAO[7].It has been shown in vivo in mice and in vivo using human platelets that TMAO causes platelet hyperreactivity and inflammation as well as foam cell formation, significantly contributing to the development of atherosclerosis [1,6,8-10]. Several preclinical and clinical studies have established a relationship between TMAO levels in blood and the risk for development of CVDs[1,4,11-13].Despite such mechanistic evidence, TMAO is solely used as a secondary marker for CVDs or atherosclerosis development in clinical practice.
Currently, the principal method to quantify TMAO levels in plasma is liquid chromatography coupled to mass spectrometry.(LC-MS)Isotope-labeled TMAO standards are used for calibration in plasma, as TMAO is endogenous in this matrix [14]. The same applies to endogenous TMA precursors such as carnitine, choline or betaine. This necessitates additional pre-analytical steps for the spiking of isotope-labeled standards into each sample, and the standards themselves increase analysis costs. In this study, we present a cost-effective,fast and sensitive method for simultaneous measurement of TMAO,carnitine,betaine and choline without the need of isotope-labeled standards. The method will facilitate the use of TMAO as a new biomarker to assess the risk of CVDs or atherosclerosis in the clinical setting. In addition, we demonstrate that only low sample volumes are needed for accurate measurements.
2. Materials and methods
2.1. Sample preparation and analysis
All samples were analyzed using an Agilent 1290 Infinity II HPLC system (Agilent, Darmstadt, Germany) coupled to an AB Sciex QTrap 6500 plus mass spectrometer(Darmstadt,Germany).First,a calibration curve was prepared by spiking different concentrations of carnitine,choline,betaine and TMAO into a phosphate-buffered saline solution containing 5%bovine serum albumin(BSA)(referred to as ‘artificial plasma matrix’). Pure TMAO, L-carnitine hydrochloride, betaine and choline chloride were purchased from Sigma-Aldrich (Darmstadt, Germany). Glipizide (Sigma-Aldrich, Darmstadt, Germany) was used as an internal standard to account for injection errors and as a quality control of the respective sample run. In addition, quality control samples (QCs) were prepared for carnitine, choline, betaine and TMAO in artificial plasma matrix.The following extraction procedure was used: 7.5 μL of a sample(calibration samples, QCs or human plasma samples) were extracted with 7.5 μL of 1% formic acid and 42.5 μL of ice-cold methanol containing 12.5 ng/mL of glipizide as internal standard for 10 min at 550 rpm on an Eppendorf MixMate®vortex mixer (Eppendorf,Hamburg,Germany).Samples were centrifuged at 8°C and 2,270 g for 5 min. Supernatants were transferred to standard HPLC-Vbottom 96-well plates (Greiner, Kremsmünster, Austria).
HPLC conditions were as follows: column, Column XP, XSelect HSS T3 (4.6 mm × 50 mm, 2.5 μm, Waters, Eschborn, Germany);temperature,30°C; injection volume, 5 μL; flow rate, 700 μL/min;solvent A,95%water +5%acetonitrile +0.1%formic acid;solvent B,95% acetonitrile +5% water +0.1% formic acid; gradient, 99% A from 0 to 0.5 min, 99%-92.2% A from 0.5 min to 0.76 min, 92.2%-92.1%A from 0.76 min to 0.78 min, 92.1% A until 0.85 min, 92.1%-92.0%A from 0.85 min to 0.90 min,92.0%-91.9%A from 0.90 min to 0.95 min,91.9%A until 1.10 min,91.9%-50%A until 2.00 min,50%-0%A from 2.00 min to 4.00 min,0%A until 5.00 min,0%-99%A from 5.00 to 6.50 min.MS conditions were as follows:multiple reaction monitoring (MRM), positive and negative modes; Q1 and Q3 masses for glipizide,carnitine,betaine,choline and TMAO shown in Table S1; peak areas of each sample and of the corresponding internal standard analyzed using MultiQuant 3.0 software(AB Sciex,Darmstadt, Germany). The internal standard served as a technical control and peak areas of internal standards were used for technical approval of HPLC-MS/MS runs.For approval of an HPLC-MS/MS run,the peak area of the respective sample had to be within±20%of the mean peak area of all measured samples.Peak areas of the internal standard were used for the calibration curve or QCs.Only runs that passed that technical control were used for further analysis. Metabolites were identified by using all ion pairs displayed in Table S1.Ion pairs used for quantification are marked with a “2” in Table S1.Peak area ratios were prepared from the respective analyte and the standard glipizide. Peak area ratios of samples were quantified using the calibration curve of the respective metabolites (i.e.,carnitine,betaine,choline or TMAO).The accuracy of the calibration curve was determined using QCs independently prepared on different days.A run was approved when the accuracy of QC values was compliant with the official European Medicines Agency guidelines for analytical determination of biomarkers (EMEA/CHMP/EWP/19221/2009 Rev.1 Corr.2).
2.2. Extraction test with water
Choline, carnitine, betaine and TMAO were either spiked into water or into the artificial plasma matrix. The extraction protocol and analysis were the same as described in Section 2.1. All four metabolites were tested at a low, middle and high concentrations.The following concentrations (in ng/mL) were used for the four metabolites: (1) carnitine: 1,000 (low), 5,000 (middle), 10,000(high);betaine:1,000(low),5,000(middle),10,000(high);choline:500 (low), 1,000 (middle), 5,000 (high); TMAO: 250 (low), 1,000(middle), 2,500 (high).
2.3. Cross-validation with mouse plasma matrix
For cross-validation experiments, metabolites were spiked into pooled plasma from CD-1 mice at concentrations of 2,500 ng/mL,5,000 ng/mL and 10,000 ng/mL to ensure that they exceeded the endogenous amounts.To rule out that effects were not attributed to a specific plasma batch,different batches were used.To determine whether spiked amounts were significantly different from the calibration curve, an ordinary one-way ANOVA was performed using Sidak's multiple comparison test using GraphPad Prism 8.4.2 software.
2.4. Stability of metabolites in human plasma samples
Human plasma samples were obtained from healthy volunteers(n =10)who had given their informed consent.Healthy volunteers had the choice to opt for venous puncture or finger prick. Plasma samples were either obtained from a finger prick (approximately 20 μL whole blood;n =1),or from venous puncture(approximately 10 mL whole blood;n =9).Whole blood samples were immediately centrifuged at 15,870 g for 10 min at 4°C to obtain plasma.Plasma samples were tested after up to six freeze-thaw-cycles (-20°C to room temperature) for their metabolite concentrations. Furthermore, metabolite levels were assessed after storage at room temperature for up to 5 days. Statistical analysis was done using an ordinary one-way ANOVA using GraphPad Prism 8.4.2 software.
3. Results
3.1. TMAO, carnitine, betaine and choline levels could be quantified simultaneously from 7.5 μL sample volume
We prepared an artificial plasma matrix consisting of albumin and phosphate-buffered saline at pH 7.4 to simulate the pH,salt and protein concentrations of plasma. We sought out to minimize the required sample volume:10 μL is the lowest sample volume so far reported in publications for the determination of TMAO, choline,carnitine and betaine[15].If sample collection from a simple finger prick is envisaged, 10 μL is still quite high, as whole blood from finger prick samples (about 20 μL) subsequently has to be processed to plasma,resulting in volume reductions of about 50%.We spiked the matrix with TMAO, carnitine, betaine and choline and prepared calibration curves as well as the independent QCs. A sample volume of 7.5 μL resulted in low variance(demonstrated by low inter-and intra-day variance of QCs)during sample processing and a broad dynamic range for the quantification of all four metabolites resulted in a robust and reproducible LC-MS/MS method(Table 1). Lower sample volumes, such as 5 μL, resulted in higher limits of quantification and a higher variance of the intra-and inter-day QCs(>±15%).Therefore,further reduction of volumes was not attempted. The correlation coefficient showed excellent linearity with R2>0.99 for each metabolite(Fig.S1A).By employing a dualcolumn switching method, the analysis of up to 240 samples per day per instrument could be achieved. As the peaks of all four metabolites were detected in MRM mode,baseline-separation was not necessary (Figs. S1B and C).

Table 1 Range of quantification, lower limit of qualification (LLOQ) and accuracy.
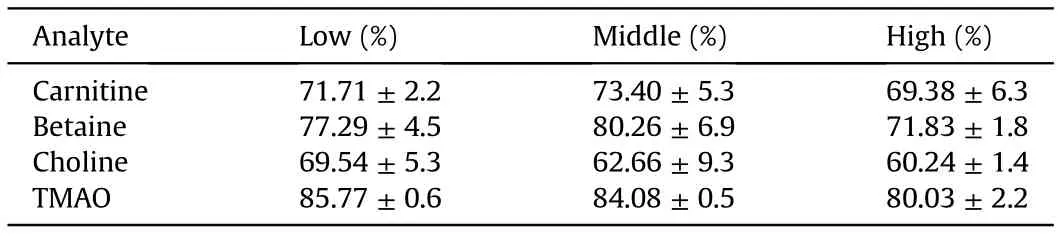
Table 2 Metabolite signals quenched by artificial matrix compared to water.
To investigate whether artificial plasma is necessary as a surrogate matrix or whether water could also be deployed,peak areas of the four metabolites at three different concentrations in water were compared to those obtained in the artificial matrix. The MS signals for carnitine, betaine and choline were quenched by up to 70%by the artificial matrix,and TMAO signals were quenched by up to 80% (Table 2). Hence, if no (isotope-labeled) internal standards are used for quantification, water cannot serve as an adequate matrix for external calibration curves, as metabolite levels would be underestimated.
3.2. Cross-validation with spiked plasma samples showed that artificial matrix was suitable for the quantification of TMAO,carnitine, betaine and choline
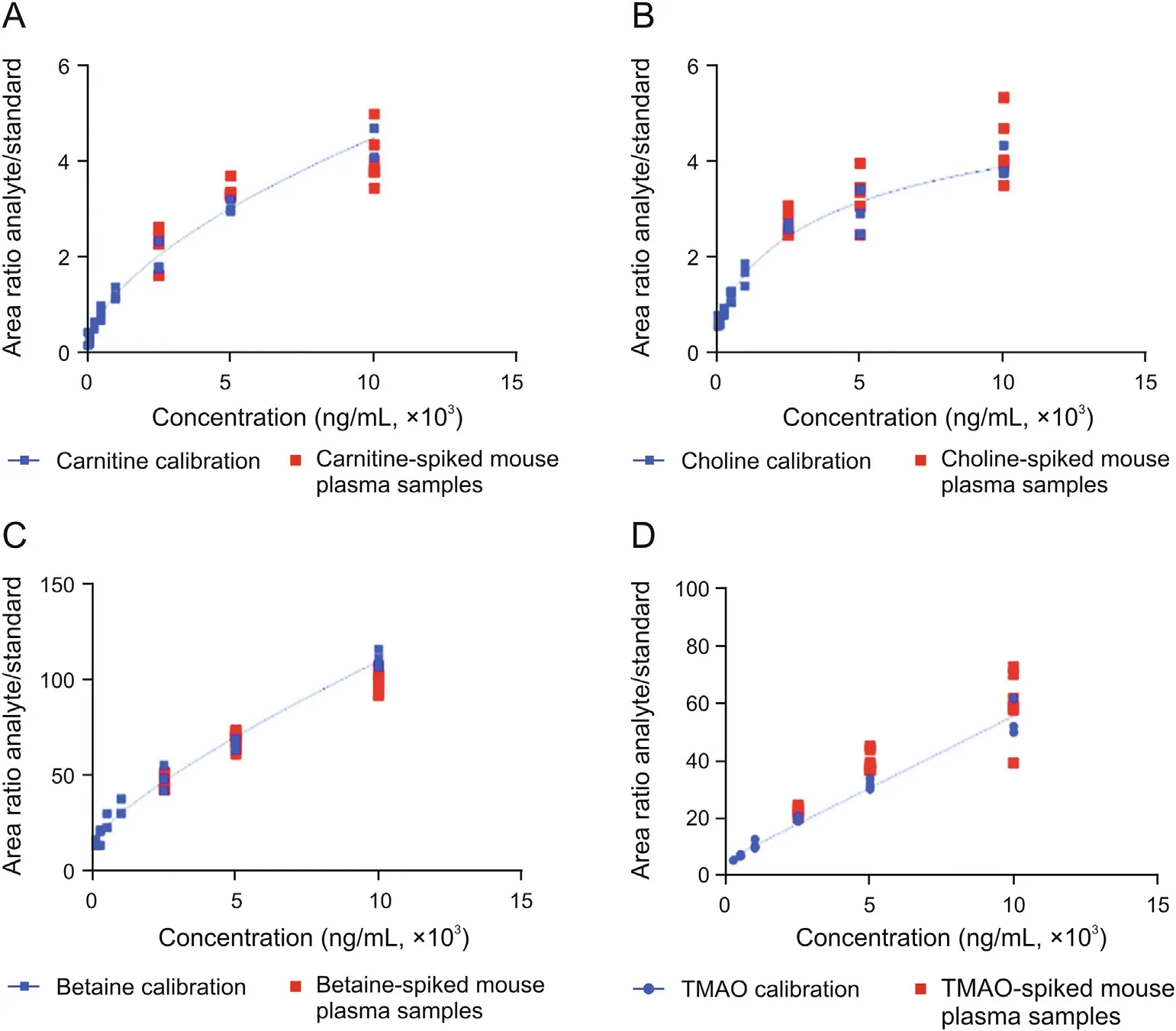
Fig. 1. Correlation of metabolite levels in mouse plasma and in artificial plasma matrix samples. (A-D) Calibration curves for (A) carnitine, (B) choline, (C) betaine and (D) trimethylamine-N-oxide (TMAO) in artificial plasma matrix. Carnitine, choline, betaine and TMAO were spiked into mouse plasma at three concentrations (2,500, 5,000 and 10,000 ng/mL) per analyte. The concentrations determined in metabolite-spiked mouse plasma samples correlate with calibration curves prepared in artificial matrix. All concentrations determined for metabolite-spiked mouse plasma samples were in the range of 80%-120%.No statistically significant difference was detected between metabolite-spiked mouse plasma samples and the respective concentrations in artificial matrix using an ordinary one-way ANOVA with Sidak's multiple comparison test.
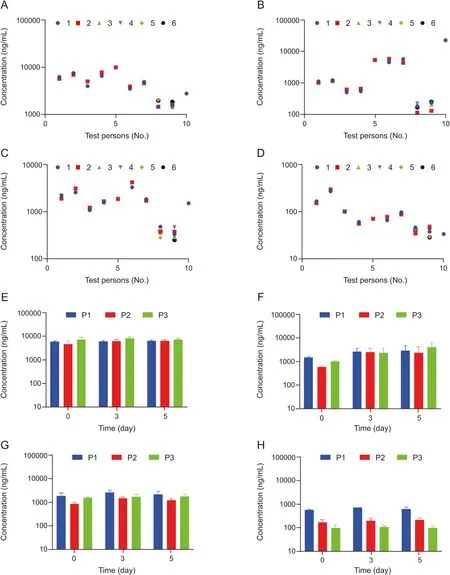
Fig.2. Stability of carnitine,choline,betaine and TMAO in plasma of healthy volunteers.(A-D)Plasma samples of ten healthy volunteers were subjected to up to six freeze-thawcycles;freeze-thaw cycle No.1(blue),No.2(red),No.3(green),No.4(purple),No.5(orange),No.6(black).Data points are partly overlaid.Plasma concentrations were determined for (A) carnitine, (B) choline, (C) betaine, and (D) TMAO. (E-H) Plasma samples of three healthy volunteers (P1, P2 and P3) were stored for up to five days at room temperature.Plasma concentrations were determined on day 0, day 3 and day 5 for (E) carnitine, (F) choline, (G) betaine, and (H) TMAO. No statistically significant differences in plasma concentrations for carnitine, choline, betaine and TMAO were detected neither for the freeze-thaw-cycles nor for the stability test up to 5 days at room temperature using an ordinary one-way ANOVA.
To prove that the artificial plasma matrix is a suitable surrogate for human plasma, the analytes were spiked into the artificial matrix at levels exceeding the reported endogenous levels for carnitine, betaine and choline by a factor of at least two and exceeding those of TMAO by a factor of at least five [1,16,17].Furthermore,different mouse plasma batches were used to secure that any observed effect was not attributed to a specific plasma batch. The concentrations measured in plasma, employing a calibration curve prepared in the artificial matrix, matched with the expected concentrations, especially for TMAO, carnitine and betaine (Fig.1). Only choline showed differences in mouse plasma batches: analysis of endogenous choline concentrations, i.e.,without choline spike, within the different mouse plasma batches revealed that endogenous choline concentrations in those two mouse plasma batches were nearly twice as high as reported in the literature[1,16,17].Thus,choline concentrations which were spiked into mouse plasma did not exceed the endogenous level by a factor of at least two in one mouse plasma batch. Finally, no statistical significant differences were detected for the analytes spiked into mouse plasma compared to those spiked into artificial plasma matrix using an ordinary one-way ANOVA test. We conclude that the validation with analyte-spiked mouse plasma samples proves that the artificial matrix is suited for the generation of external calibration curves for LC-MS/MS analysis, thereby eliminating the need for isotope-labeled internal standards.
3.3. Stability tests of TMAO, carnitine, betaine and choline
The plasma levels of TMAO,carnitine,betaine and choline were determined simultaneously in plasma obtained from either venous puncture or finger prick blood from healthy volunteers (Figs.2A-D). Concentrations of all four metabolites were in the same range as previously described for healthy volunteers [1,6,14,16,18].Moreover, we did not observe significant differences in concentrations between samples from either a finger prick or venous puncture. This demonstrates that a plasma sample from a finger prick is sufficient to determine levels of all four metabolites.Furthermore, we show that our method is suitable for measuring‘real-life’ samples from humans.
Next,we wanted to assess the stability of the four metabolites in‘real-life’samples from humans.Up to six freeze-thaw cycles of the samples had no influence on metabolite concentrations as no statistically significant difference was detected for concentrations after several freeze-thaw cycles (Figs. 2A-D). Storage at room temperature for up to 5 days had no influence on the concentrations of carnitine, betaine or TMAO, either. It seems that the concentrations of choline increased slightly over time.However,using an ordinary one-way ANOVA, no statistically significant difference for the concentrations of all four metabolites on the days 0, 3 or 5 was determined (Figs. 2E-H). Hence, all four metabolites were stable when appropriately stored at-20°C, whereas storage at room temperature could cause misleading results for choline plasma concentrations. Furthermore, we provide proof-of-concept that our method is capable of obtaining similar results as the current standard method using isotope-labeled standards.
4. Discussion
TMAO has emerged as a promising marker for prognosis and monitoring of CVDs or atherosclerosis [2]. The current standard method uses isotope-labeled standards of TMAO and its precursors in plasma [14,19-21]. Although label-free methods have been published in the recent years deploying different approaches for a suitable surrogate matrix[22,23],isotope-labeled standards are still the method of choice for testing patient cohorts. We provide a more simple standard method to measure the plasma levels of TMAO and its precursors carnitine,betaine and choline for the validation of their full medical utility in diverse populations,in order to facilitate their wider analysis as possible biomarkers [12,13].We have developed a robust method with good reproducibility using column-switching that is fast and requires a minimal sample volume of just 7.5 μL. In comparison to recently published label-free methods, our method needs far less sample volume with the same quantification range and slightly lower levels of detection for all four metabolites[14,15,23,24].In addition,we used a fast sample preparation procedure compared to previously reported label-free methods[24]so that samples can be analyzed within several hours. We demonstrated that an artificial matrix sufficiently mimics human plasma matrix for the analysis of the four metabolites. Thus, external calibration is possible and isotope-labeled standards are not needed. Furthermore, we provide evidence that metabolite levels in human ‘real-life’ plasma samples are determined with high precision, and that the plasma levels of carnitine,betaine,choline and TMAO are comparable to those in the literature[14,16,18].
Moreover,we demonstrated that a sample volume of only 7.5 μL can also be obtained via a finger prick that is post-processed to plasma.This low sample volume has not been reported in previous studies [23,24]. Recently, the first prototype device has been developed to obtain 10 μL of plasma from a finger prick within 10 min [25]. Combination of that passive plasma filtration device with our fast LC-MS/MS method paves the way for patient-centric monitoring of the risk of CVDs or atherosclerosis in clinical routine.
The runtime currently allows analyzing up to 240 samples per day per instrument, including sample pre-treatment. It can be further shortened by optimized gradients: the peaks for all four metabolites are well separated and eluted within 1 min. By changing the internal standard glipizide to an earlier eluting standard, method runtimes could be in principle reduced to 2-2.5 min, which would translate to an increase in throughput to around 570 samples per day per instrument.
The current choice of glipizide as an internal standard is based on the fact that glipizide has no German market authorization as an antidiabetic drug. Thus, it will not be found in German patient samples. For screening large cohorts from different countries, glipizide needs to be replaced by a different exogenous standard that fulfills the quality criteria; a related option could be the ‘outdated’sulfonylurea carbutamide. A proof-of-concept that our method is suitable for screening large cohorts in Germany for cardiovascular risk assessment will be reported soon.
5. Conclusion
In summary, we provide a fast and cost-effective LC-MS/MS method with very low sample volume for the simultaneous detection of TMAO and its precursors carnitine, betaine and choline. This method in combination with a finger-prick device facilitates the establishment of TMAO and its precursors as biomarkers in clinical practice, thereby enabling control of disease progression for atherosclerosis and CVDs. Physicians can directly determine patients at risk, control disease progression, and make the successful dietary interventions with just a finger prick.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors thank Dr. Friederike Klein for providing human plasma samples.Katharina Rox received support from the German Centre for Infection Research (DZIF, TTU 09.710). The work was supported by the Helmholtz Association's Initiative on Aging and Metabolic Programming (AMPro).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2021.03.007.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Plasma-metabolite-based machine learning is a promising diagnostic approach for esophageal squamous cell carcinoma investigation
- Reducing SARS-CoV-2 pathological protein activity with small molecules
- UHPLC-MS/MS analysis of cAMP and cGMP in rat plasma as potential biomarkers of Yin-Yang disharmony in traditional Chinese medicine
- Liquid chromatography-mass spectrometry method for the quantification of an anti-sclerostin monoclonal antibody in cynomolgus monkey serum
- Simultaneous determination of fourteen components of Gumiganghwal-tang tablet in human plasma by UPLC-ESI-MS/MS and its application to pharmacokinetic study
- Development of the general chapters of the Chinese Pharmacopoeia 2020 edition: A review