Simultaneous determination of fourteen components of Gumiganghwal-tang tablet in human plasma by UPLC-ESI-MS/MS and its application to pharmacokinetic study
2021-09-14SeungHyunJeongJiHunJngGukYeoLeeSeungJungYngHeYoungChoYongBokLee
Seung-Hyun Jeong , Ji-Hun Jng , Guk-Yeo Lee , Seung-Jung Yng , He-Young Cho ,Yong-Bok Lee ,*
a College of Pharmacy, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju, 61186, Republic of Korea
b National Development Institute of Korean Medicine, 288 Udeuraendeu-gil, Anyang-myeon, Jangheung-gun, Jeollanam-do, 59338, Republic of Korea
c Department of Oriental Rehabilitation Medicine, Dongshin University Gwangju Korean Medicine Hospital,141 Wolsan-ro, Nam-gu, Gwangju, 61619,Republic of Korea
d College of Pharmacy, CHA University, 335 Pangyo-ro, Bundang-gu, Seongnam-si, Gyeonggi-do,13488, Republic of Korea
Keywords:Gumiganghwal-tang Fourteen bioactive ingredients Simultaneous analysis UPLC-ESI-MS/MS Pharmacokinetics Clinical study
ABSTRACT Gumiganghwal-tang is a traditional herbal medicine widely used for its anti-inflammatory, analgesic,and antipyretic effects. However, the safety and efficacy of its active ingredients based on an in vivo pharmacokinetic (PK) study have yet been investigated. We have established a sensitive and accurate UPLC-ESI-MS/MS method and conducted a PK study on 14 constituents of Gumiganghwal-tang through human plasma analysis. Analytical conditions were optimized according to the physicochemical properties of the 14 compounds to facilitate efficient separation and eliminate overlap or interference between peaks.KINETEX-C18 and Inertsil-C8 columns were used as UPLC stationary phases,and acetonitrile and aqueous formic acid were used as mobile phases. All the analytes were quantified with a triple quadrupole mass spectrometer using electrospray ionization in multiple reaction monitoring mode.The chromatograms of 14 bioactive compounds showed excellent elution and sensitivity,and each peak was selectively separated and quantified without interference with each other or impurities.The established analytical method was based on international guidelines and was successfully used to perform PK studies of 14 herbal ingredients in humans after oral administration with Gumiganghwal-tang tablets. The oral absorption of most active components of Gumiganghwal-tang was relatively rapid and remained considerably long in the body to be quantified in plasma up to 48 h after administration.
1. Introduction
Gumiganghwal-tang is known as Kumi-Kyokatsu-to in Japan and Jiuweiqianghuo-tang in China [1]. The multiple names of Gumiganghwal-tang are attributed to its long history of clinical use in East Asian countries [2]. Gumiganghwal-tang is a traditional herbal medicine with anti-inflammatory,analgesic,and antipyretic effects [3]. Therefore, it is prescribed for common cold, inflammatory diseases, and pain [4]. Gumiganghwal-tang comprises nine crude herbal medicines derived from Angelica koreana Maxim.,Saposhnikovia seseloides Kitag.,Cnidium officinale Makino.,Angelica dahurica (Hoffm.) Benth. & Hook.f. ex Franch. & Sav., Atractylodes japonica Koidz.ex Kitam.,Scutellaria baicalensis Georgi,Rehmannia glutinosa (Gaertn.) DC., Asiasarum sieboldii Miq., and Glycyrrhiza uralensis Fisch[1].The major constituents of each herbal medicine interact via a complex mechanism resulting in clinical manifestations [5]. Various active ingredients in Gumiganghwal-tang may also have synergistic therapeutic effects with other herbal medicines [6]. This synergistic effect is probably attributed to the multiple active ingredients in herbal medicine. And traditional herbal medicines exhibit strong therapeutic effects with relatively high safety and low toxicity in vivo[7].Therefore,research and interest in traditional herbal medicines have continued from the past to the present time. In clinical practice, prescriptions of herbal medicine for related diseases are increasing [5]. However, studies that evaluate its safety and efficacy based on in vivo pharmacokinetic (PK)information of its main ingredients are lacking, although herbal medicines are gaining increasing attention and clinical usage[8].
In the case of Korea,there is a growing need to adjust or expand insurance coverage by the National Health Insurance for herbal prescriptions.Among them,Gumiganghwal-tang is the fourth most commonly prescribed herbal medicine [9]. Although Gumiganghwal-tang has been used in humans for a long time and its efficacy has been recognized empirically,accurate evaluation of its safety and efficacy based on clinical PKs is critical for routine clinical application.Unfortunately,despite the frequent clinical use of Gumiganghwal-tang, accurate PK information has not been reported.
On the other hand, we have already reported the development of an analytical assay involving commercial Gumiganghwal-tang tablets in a previous study [10]. However, the reported assay [10]was only useful for in vitro analysis for formulation and not for the analysis of biological samples. Therefore, for the PK research, it is necessary to develop and validate new analytical method based on the previous method.
The purpose of this study was to perform PK studies using analytical methods developed for the simultaneous determination of 14 main ingredients of Gumiganghwal-tang based on human plasma analysis. To the best of our knowledge, studies that use UPLC-ESI-MS/MS to determine pharmacologically active ingredients or estimate their clinical PK parameters in humans orally administered with Gumiganghwal-tang have yet been reported.
In this study, the 14 major herbal components we selected as active ingredients for Gumiganghwal-tang's PK study were as follows: baicalin, baicalein, 18-beta-glycyrrhetinic acid, glycyrrhizin,xanthotoxol,imperatorin,aucubin,catalpol,ferulic acid,notopterol,methyl eugenol, oxypeucedanin, atractylenolide III, and atractylenolide I. These 14 components included the main ingredients contained in each of the nine crude herbal medicines of Gumiganghwal-tang. As a brief description of each selected ingredient, baicalin and its metabolite baicalein are major active ingredients of S. baicalensis [11]. Glycyrrhizin and its metabolite 18-beta-glycyrrhetinic acid are major active ingredients of G. uralensis [12]. Imperatorin and its metabolite xanthotoxol are major active ingredients of S.divaricata[13].Although baicalein,18-beta-glycyrrhetinic acid, and xanthotoxol are major metabolites in vivo, they may occur in herbal medicines together with their prodrugs or may be produced by heat or other external stimuli during manufacture and formulation as suggested in previous reports [12-14]. These results were confirmed by our recent study analyzing the major constituents in Gumiganghwal-tang tablet[10].Oxypeucedanin is a major active ingredient of A.dahurica[15].Atractylenolides I and III are major active ingredients of A.japonica[16]. Aucubin and catalpol are major active ingredients of R. glutinosa [17]. Methyl eugenol is a major active compound of A. sieboldii [18]. Ferulic acid is a main active component of C. officinale [19] and notopterol is the key active ingredient in O. koreanum [20]. Fig. S1 presents the structures of these 14 key bioactive ingredients.
2. Experimental
2.1. Reagents and chemicals
Gumiganghwal-tang tablets were supplied by Kyung-bang Pharmaceutical Company (Incheon, Republic of Korea). Reference standards for baicalin (purity ≥98.2%; Cas No. 21967-41-9), glycyrrhizin (purity ≥ 99.3%; Cas No. 1405-86-3), notopterol(purity ≥98.9%;Cas No.88206-46-6),xanthotoxol(purity ≥98.4%;Cas No. 2009-24-7), oxypeucedanin (purity ≥98.1%; Cas No. 737-52-0), methyl eugenol (purity ≥98.3%; Cas No. 93-15-2), and catalpol (purity ≥98.0%; Cas No. 2415-24-9) were obtained from ChemFaces (Wuhan, Hubei Province, China). Reference standards for 18-beta-glycyrrhetinic acid(purity ≥99.0%;Cas No.471-53-4),baicalein (purity ≥ 99.0%; Cas No. 491-67-8), aucubin(purity ≥98.0%;Cas No.479-98-1),ferulic acid(purity ≥99.0%;Cas No. 1135-24-6), imperatorin (purity ≥98.0%; Cas No. 482-44-0),atractylenolide I (purity ≥98.0%; Cas No. 73069-13-3), and atractylenolide III(purity ≥98.0%;Cas No.73030-71-4)were purchased from Sigma-Aldrich (St. Louis, MO, USA). Reference standards for repaglinide (purity ≥99%; Cas No. 135062-02-1) and celecoxib(purity ≥99%;Cas No.169590-42-5)as internal standards(IS)were also supplied by Sigma-Aldrich (St. Louis, MO, USA). Water, methanol,and acetonitrile of LC-MS grade were supplied by DUKSAN Inc(Ansan, Republic of Korea). Formic acid of LC-MS grade and ammonium formate were provided by Thermo Fisher Scientific(Waltham,MA,USA).Acetic acid of HPLC grade was purchased from Tokyo Chemical Industry (Tokyo, Japan). All chemicals used in this study conformed to the highest HPLC grade or quality available.
2.2. Composition of Gumiganghwal-tang tablet
We adopted the method reported previously[10]to evaluate the composition of 14 main ingredients in Gumiganghwal-tang tablets.Briefly, more than 20 tablets of Gumiganghwal-tang were finely ground with a tablet mill (Yuhan Medical Co., Daegu, Republic of Korea). The ground powder was then classified according to particle size through a sieve analyzer (Fritsch, Idar-Oberstein, Germany), and 3.2 g of the powder was passed through a 45 mesh(<355 μm)sieve,followed by ultrasonic extraction with 100 mL of 80% (V/V) aqueous methanol at 40 ± 2.5°C water bath for 90 min.The extracted solution was filtered through a 0.45 μm microporous membrane and then adjusted to the original volume by adding 80%(V/V) methanol.The sample was finally centrifuged at 10,000 g for 5 min. The 14 major constituents of this extract were simultaneously quantified using LC-MS/MS as reported previously [10].Amounts of baicalin, baicalein,18-beta-glycyrrhetinic acid, glycyrrhizin, xanthotoxol, imperatorin, aucubin, catalpol, ferulic acid,notopterol,methyl eugenol,oxypeucedanin,atractylenolide III,and atractylenolide I in the extract were 10146.169 ± 948.328,1041.797 ± 100.243, 14.345 ± 4.049, 997.281 ± 124.127,2.769 ± 0.583, 0.135 ± 0.054, 1.125 ± 0.305, 0.793 ± 0.100,8.896 ± 0.432, 0.073 ± 0.010, 0.072 ± 0.029, 128.685 ± 25.353,34.099 ± 5.220 and 3.236 ± 0.396 μg/g, respectively.
2.3. Instrumentation and analytical conditions
2.3.1. Instrumentation
Shimadzu Nexera-X2 Series UPLC system (Shimadzu, Kyoto,Japan) coupled with a Shimadzu-8040 mass spectrometer with a DGU-20A degassing unit, an SIL-30AC autosampler, and an LC-30AD pump was used for simultaneous quantitative analysis of 14 key ingredients in human plasma samples.
2.3.2. Chromatographic conditions
Optimized chromatographic separation of 12 herbal compounds including baicalin, xanthotoxol,18-beta-glycyrrhetinic acid, baicalein, glycyrrhizin, imperatorin, atractylenolide I, ferulic acid, oxypeucedanin, atractylenolide III, notopterol, and methyl eugenol,was performed with a KINETEX-C18column(50 mm×2.1 mm i.d.,1.7 μm particle size; Phenomenex Inc., Torrance, CA, USA). The mobile phase was performed with 0.1%(V/V)formic acid in water as mobile phase A and 100%acetonitrile as mobile phase B with a flow rate of 0.3 mL/min and a gradient elution. The elution profile was optimized as follows: 0-0.5 min,10% B; 0.5-1.5 min,10%-60% B;1.5-2.5 min, 60% B; 2.5-3.5 min, 60%-90% B; 3.5-4.25, 90% B;4.25-4.26 min, 90%-10% B; and 4.26-5.0 min, 10% B. Optimized chromatographic separation of two components including catalpol and aucubin was performed with an Inertsil-C8-3 column(100 mm × 2.1 mm i.d., 2.0 μm particle size; GL Sciences, Tokyo,Japan).The mobile phase was performed with 100%acetonitrile as mobile phase B and 10 mM aqueous ammonium formate supplemented with 0.01%(V/V)formic acid as mobile phase A with a flow rate of 0.2 mL/min and gradient elution. The elution profile was optimized as follows:0-0.5 min,10%B;0.5-1.25 min,10%-90%B;1.25-3.5 min,90%B;3.5-3.51 min,90%-10%B;and 3.51-5.0 min,10% B. For all analytes, the column and autosampler tray temperatures were maintained at (40 ± 2.5) and(15 ± 1)°C, respectively.The total analytical run time was 5 min per sample. The injection volume was 5 μL for all analyses.
2.3.3. Mass spectrometry conditions
Detection and quantification of all analytes were performed using a triple quadrupole mass spectrometer employing the multiple reaction monitoring(MRM)mode.Analytical procedures were evaluated with positive and negative electrospray ionization (ESI)methods, respectively, depending on materials. Positive ESI was used for the analysis of methyl eugenol, xanthotoxol, atractylenolide I, glycyrrhizin, imperatorin, atractylenolide III, notopterol,18-beta-glycyrrhetinic acid, oxypeucedanin, and IS (repaglinide),while negative ESI was used for aucubin, ferulic acid, baicalein,baicalin, catalpol, and IS (celecoxib). Electrospray ion source was operated with polarity switching between positive and negative ion modes in a single run. Optimized parameters of ESI interface were set as follows: ion source temperature, 250°C; desolvation temperature,400°C;capillary voltage,1.88 kV;nebulizing gas flow,3 L/min;and drying gas flow,15 L/min.Dwell time of all ingredients was 100 ms.Other optimized MS/MS parameters of each ingredient are presented in Table S1.
2.4. Preparation of quality control (QC) samples and calibration curve
Each standard of baicalin,baicalein,18-beta-glycyrrhetinic acid,glycyrrhizin, xanthotoxol, imperatorin, aucubin, catalpol, ferulic acid, methyl eugenol, oxypeucedanin, notopterol, atractylenolide III, atractylenolide I, and IS (repaglinide and celecoxib) was weighed accurately and dissolved in methanol at 1 mg/mL to prepare a stock solution before obtaining each working solution.These stock solutions were stored at-20°C.Standard working solutions of glycyrrhizin (1-5000 ng/mL), 18-beta-glycyrrhetinic acid(5-5000 ng/mL), imperatorin (1-5000 ng/mL), xanthotoxol(5-5000 ng/mL),oxypeucedanin(5-5000 ng/mL),methyl eugenol(10-5000 ng/mL),baicalin(5-5000 ng/mL),baicalein(5-5000 ng/mL), atractylenolide I (1-5000 ng/mL), atractylenolide III(5-5000 ng/mL), notopterol (1-5000 ng/mL), ferulic acid(5-5000 ng/mL), aucubin (5-6000 ng/mL), catalpol (5-6000 ng/mL), and IS (100 ng/mL for repaglinide, 500 ng/mL for celecoxib)were obtained from their standard stock solutions by diluting with 100% methanol. Here the 14 analytes were mixed using stock solutions and stepwise diluted with methanol to prepare the working solutions. Calibration standards were obtained by adding each diluted working solution into blank human plasma samples obtained from six different individuals to determine the final concentrations of glycyrrhizin, imperatorin, atractylenolide I, and notopterol (ranging from 0.1 to 500 ng/mL), xanthotoxol,18-betaglycyrrhetinic acid,oxypeucedanin,baicalein,baicalin,ferulic acid,and atractylenolide III (ranging from 0.5 to 500 ng/mL), methyl eugenol (ranging from 1 to 500 ng/mL), aucubin, and catalpol(ranging from 0.5 to 600 ng/mL). To determine the precision and accuracy of analysis,QC samples were similarly prepared by mixing the 14 compounds at four different levels: (1) glycyrrhizin, imperatorin,atractylenolide I,and notopterol of 0.1,2,80,and 400 ng/mL;(2) xanthotoxol, 18-beta-glycyrrhetinic acid, oxypeucedanin, baicalein,baicalin,ferulic acid,and atractylenolide III of 0.5,2,80,and 400 ng/mL;(3)methyl eugenol of 1,5,80,and 400 ng/mL;and(4)aucubin and catalpol of 0.5,2,80,and 500 ng/mL.QC samples were prepared or evaluated and calibrated on the same day of analysis.
2.5. Sample preparation
Human plasma samples, calibration standards, and QC samples were extracted by liquid-liquid extraction(LLE)using ethyl acetate supplemented with 1% (V/V) acetic acid. Protein was precipitated via protein-precipitation(PP)method using methanol.A 10 μL of IS solution (repaglinide of 100 ng/mL, celecoxib of 500 ng/mL) was mixed with a human plasma sample of 100 μL, followed by the addition of 1000 μL ethyl acetate supplemented with 1%(V/V)acetic acid-methanol (2:8, V/V), vortexed for 5 min and centrifuged at 13,500 g for 5 min. Then,1000 μL of the supernatant organic layer was dried gently with a centrifugal evaporator using nitrogen gas at 25°C. The dried matter was reconstituted with 50 μL of mobile phase and vortexed for 5 min. After centrifugation at 13,500 g for 5 min, 5 μL of aliquot was injected for UPLC-ESI-MS/MS analysis.
2.6. Method validation
The newly developed UPLC-ESI-MS/MS method for in vivo assay was fully validated in terms of sensitivity, selectivity, linearity, accuracy, precision, matrix effect, recovery, stability, carryover, and incurred-sample-reanalysis(ISR) in accordance with the Guidance for Industry: Bioanalytical Method Validation by Food and Drug Administration[21].
2.6.1. Sensitivity and selectivity
Selectivity was investigated to confirm the influence of endogenous compounds located in the closed retention time of the analytes. Blank plasma, zero plasma (plasma spiked with IS),plasma spiked with all 14 herbal compounds (lower limit of quantitation(LLOQ)and upper limit of quantitation(ULOQ)for the 14 compounds), and human plasma samples collected after oral administration of Gumiganghwal-tang tablets (9.6 g) to Korean subjects were used to demonstrate selectivity. Sensitivity of the method was expressed as the LLOQ determined as the lowest concentration of standard samples with a signal-to-noise ratio of at least 10:1 in accordance with an acceptable precision of less than 20%and accuracy within±20%.It was evaluated using five replicate samples.
2.6.2. Linearity and LLOQ
Calibration curves were drawn at seven calibration points by linear regression with a weighting factor of 1/concentration2.Linearity was determined by plotting the analyte/IS peak area ratio versus theoretical concentration of the analyte.A linear calibration equation with its correlation coefficient (r2) was presented. Linearities for 14 analytes were determined with a series of calibration standards in the range of 0.1-500 ng/mL(glycyrrhizin,imperatorin,atractylenolide I, and notopterol), 0.5-500 ng/mL (xanthotoxol,atractylenolide III, oxypeucedanin, 18-beta-glycyrrhetinic acid,baicalin, baicalein, and ferulic acid), 0.5-600 ng/mL (aucubin and catalpol), or 1-500 ng/mL (methyl eugenol) in human plasma. A straight-line regression equation was determined with r2of 0.99.LLOQ was the lowest concentration within the calibration range as mentioned above (Section 2.6.1). When the measured sample concentrations were above the upper limit of the standard curve(especially for the measurement of baicalin concentration),samples were diluted with blank human plasma and reanalyzed.The dilution integrity had been proved by the standard analytes,whose concentration was above the ULOQ, spiked with blank plasma.That is,the effect of sample dilution was evaluated at 40 μg/mL of baicalin. This sample (as 40 μg/mL of baicalin) was diluted 100-fold with blank human plasma and analyzed according to the same protocol used for plasma samples (Section 2.5). Dilution integrity was evaluated in five replicates (n =5).
2.6.3. Accuracy and precision
Intra-day accuracy and precision were determined by analyzing QC samples at four concentration levels for each analyte at five different times on the same day (n =5). Inter-day assessments(of accuracy and precision) were similarly conducted on five consecutive days (n =5). The concentration of each QC sample was determined using freshly prepared calibration standards.Accuracy was evaluated by ensuring that the mean value did not exceed 15%of the nominal concentration except for LLOQ which did not exceed 20%. Precision was determined by calculating the coefficient of variation (CV) for the analysis of QC samples. CV of precision for each concentration level did not deviate by more than±15%except for LLOQ, which has a limit of 20%.
2.6.4. Matrix effect and recovery
Recoveries of the 14 herbal compounds were evaluated for QC samples at low, medium, and high concentrations with five replicates.The extraction recoveries of the 14 components from human plasma samples were assessed by comparing detector (MS/MS)responses for extracted samples with those of samples added at the same concentration after extracting blank plasma.Recoveries of IS were also evaluated at working concentration (10 ng/mL for repaglinide, 50 ng/mL for celecoxib) similarly. Additionally, the matrix effect was determined by comparing the peak area of analyte postextraction from blank plasma with absolute standard of the same.
2.6.5. Stability
He said I have something to tell you, I have a one month old daughter . I asked do you still have ties with the mother? He then offered that they had been separated for the duration of the pregnancy3() and that he was sent away when he attempted to visit the hospital shortly after she was born.
The stability of 14 herbal metabolites in human plasma samples was determined under various conditions: short-term stability,long-term stability,freeze-thaw stability,and autosampler stability as post-preparative.Two different concentrations(low and high)of QC samples were analyzed in all stability tests.Short-term stability test was carried out by maintaining QC samples at 25°C for 24 h.Long-term stability was determined by analyzing QC samples frozen at-80°C for 4 weeks. To conduct the freeze-thaw stability test, QC samples were stored at-80°C for 24 h and then thawed completely at 25°C. The cycle was repeated and the analysis was performed after the third cycle. In addition, QC samples were placed in the autosampler at 15°C for 24 h for post-preparative stability test. Stabilities of stock solutions of 14 herbal metabolites and IS were assessed by determining the concentrations of analytes after storage at-20°C for 4 weeks. Samples were considered stable if the mean peak area at each level was within±15% of the sample nominal concentration and the precision was less than 15% (n=5).
2.6.6. Carryover
Carryover was conducted to determine whether the IS or analytes remaining in the analytical instrument during the subsequent analysis of the sample affected quantification and analysis of material in the next sample injection.A carryover test was conducted by injecting a blank sample after injecting the maximum concentration sample of each analyte. In this blank sample, each analyte peak was less than 20% of the LLOQ peak.
2.6.7. Incurred sample reanalysis (ISR)
ISR was carried out to ensure reproducibility of the newly developed method for the analysis of 14 herbal compounds. It was performed on different days using batches different from the initial analysis within the time period during which sample stability was ensured.Samples near the elimination phase in the PK profile of the analyte and the maximum plasma concentration (Cmax) were selected(10%or more of the total analyzed samples).Twenty human plasma samples were selected and the results were compared with initially analyzed values. The acceptance criterion was that twothirds of repeat values were within 100% ± 20% of original values.ISR deviation was calculated according to the following equation:

2.7. Application to PK study
This clinical trial was performed in accordance with the revised Declaration of Helsinki for biomedical research involving human subjects and Recommendations for Good Clinical Practice. In addition, the study protocol was approved by the Institutional Review Board of Dongshin Oriental Medicine Hospital(Gwangju,Republic of Korea;IRB approval number:2018-11).Twelve healthy Korean men(21-32 years of age; body weight, 60.3-81.6 kg; height,170.0-189.0 cm)were recruited for this clinical trial.All participants provided written informed consent and underwent a physical examination, medical history taking, and laboratory tests prior to full clinical trial participation. Each participant was sufficiently healthy for inclusion. All participants fasted for more than 10 h before receiving the tablet and fasted again for 4 h thereafter. In addition,they were advised to avoid foods or drinks containing caffeine or xanthine that could affect clinical trial results. Each participant received Gumiganghwal-tang 12 tablets(9.6 g,at the maximum oral dose) with 240 mL of water. Blood samples (6-7 mL each) were collected from the forearm vein before administration (0 h) and at 0.17, 0.33, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h after oral administration.They were placed into BD Vacutainer®spray-coated EDTA tubes (Thomas Scientific, Swedesboro, NJ, USA) and immediately centrifuged at 10,000 g for 10 min at 4°C.Each plasma sample was placed in a polyethylene tube and stored at-80°C before analysis. Time to reach Cmax(Tmax) and Cmaxwere individually determined based on the plasma concentration-time curve. Area under the curve from 0 to infinity (AUC0-∞) was integrated by a linear trapezoidal rule to the final measured concentration(Clast)and extrapolated to infinity by adding the area from Clastto infinity(Clast/k), in which k denotes the elimination rate constant at terminal phase. Half-life (t1/2) was determined as 0.693/k and volume of distribution(Vd/F)was determined as dose/k·AUC0-∞,where F refers to its bioavailability at oral administration.Systemic clearance(CL/F)was determined by dividing each dose of the analyte by AUC0-∞.All PK parameters were determined via noncompartmental analysis using WinNonlin®program, version 8.1 (Pharsight, a Certara™Company,CA,USA).
3. Results and discussion
3.1. Method development
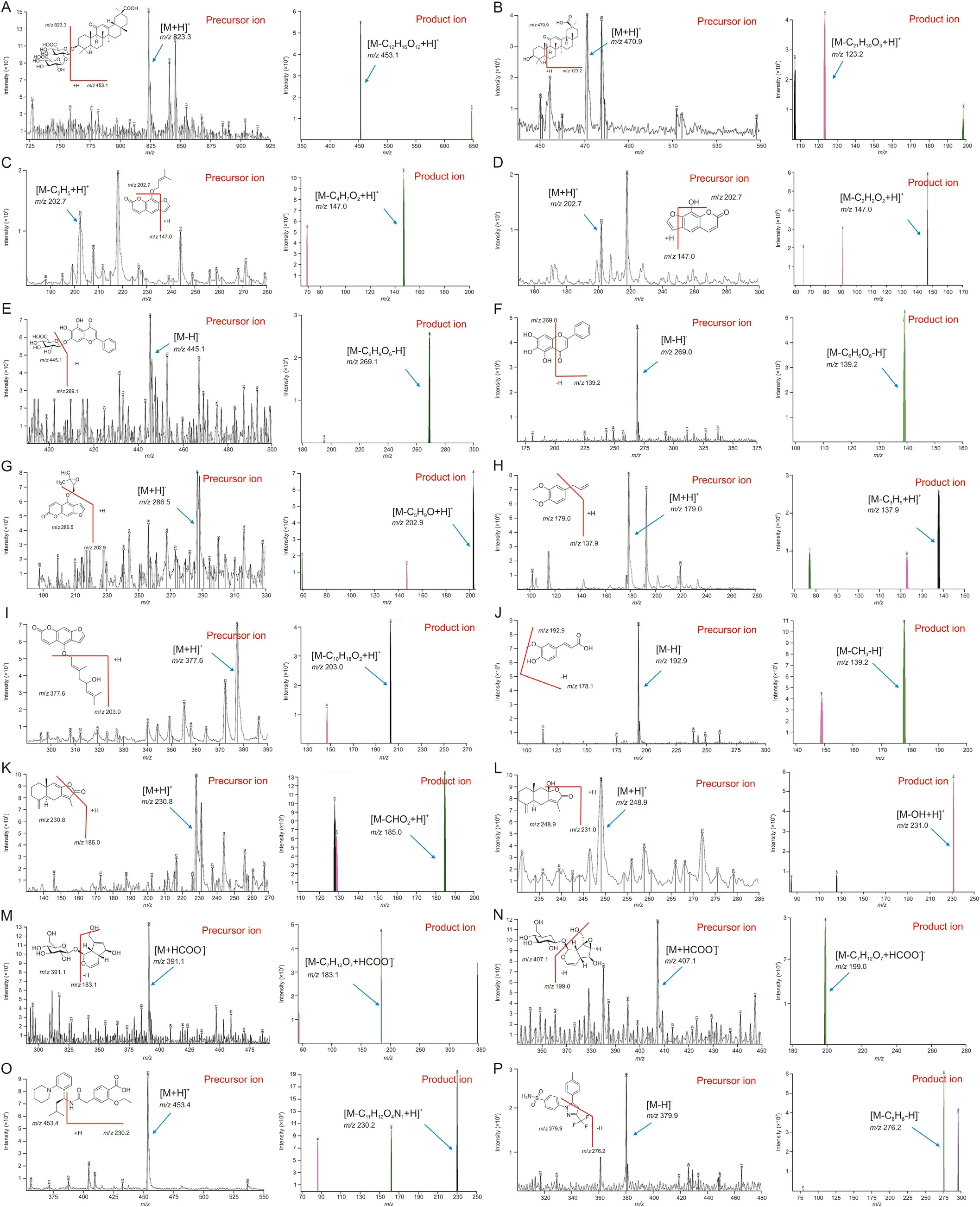
Fig.1. Precursor and product ion mass spectra of(A)glycyrrhizin,(B)18-beta-glycyrrhetinic acid,(C)imperatorin,(D)xanthotoxol,(E)baicalin,(F)baicalein,(G)oxypeucedanin,(H)methyl eugenol,(I)notopterol,(J)ferulic acid,(K)atractylenolide I,(L)atractylenolide III,(M)aucubin,(N)catalpol,and(O)repaglinide and(P)celecoxib as internal standards(IS).
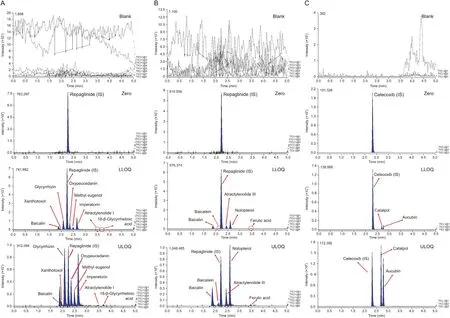
Fig. 2. Representative MRM chromatograms of blank plasma, zero plasma containing internal standard (IS), blank plasma containing LLOQ of 14 herbal compounds, and blank plasma containing ULOQ of 14 components. (A) Simultaneous analysis of glycyrrhizin, 18-beta-glycyrrhetinic acid, imperatorin, xanthotoxol, oxypeucedanin, methyl eugenol,baicalin, and atractylenolide I; (B) simultaneous analysis of baicalin, baicalein, notopterol, ferulic acid, and atractylenolide III; (C) simultaneous analysis of aucubin and catalpol.
Based on the previous research [10], a sensitive and accurate UPLC-ESI-MS/MS method was established in this study and mass spectrometric parameters were optimized for simultaneous determination of 14 ingredients in human plasma. The chromatographic and mass spectrometry conditions associated with the analysis were similar to those of our previous study [10].However, the previous study only evaluated the concentration of major components in commercial formulations,and therefore,was not readily applicable to the analysis of biological samples. As a result, further development of methods for plasma sample preparation and validation of established biological sample assays was required.
Product ion mass spectra of 14 ingredients and IS were obtained in scan mode after the individual standard solution was injected into the mass spectrometer. Signal intensity of (M+HCOO-)-ions was substantially higher than that of (M-H)-ions for catalpol and aucubin, while the signal intensities of(M-H)-ions for ferulic acid,baicalein, baicalin, and celecoxib as IS were significantly higher in negative ESI mode. The results were attributed to the presence of several hydroxyl, carboxyl, or sulfonamide groups in the analytes,which could be easily ionized in the negative ESI mode.However,the signal intensities of(M+H)+ions were the most abundant for methyl eugenol, atractylenolide III, notopterol, 18-beta-glycyrrhetinic acid,xanthotoxol, glycyrrhizin, atractylenolide I, oxypeucedanin, and repaglinide as IS in positive ESI mode. Furthermore, the signal intensity of(M-C2H5+H)+ions was stronger than that of(M+H)+ions for imperatorin.Therefore,(M-C2H5+H)+was determined as a precursor ion.Although imperatorin exhibited the same mass spectrum as metabolite xanthotoxol, it did not interfere with quantitation because peak separation on the column was complete, as reported previously [22]. Fig.1 presents relevant mass spectra. To accurately determine the 14 ingredients simultaneously,we applied conditions in which the peak retention time of each component did not overlap with each other for complete separation. As a result, 14 different ingredients were analyzed simultaneously, two of which required different columns (Inertsil C8-3 column) due to differences in intrinsic polarity of materials. The remaining 12 ingredients were divided into two to avoid overlap in retention time between analytes with the same column (KINETEX core-shell C18column) and analytical conditions. As a result, we completely eliminated the possibility of interference and cross-talk when analyzing various materials. In addition, IS solution addition was used to minimize volume error. Repaglinide and celecoxib were used as IS because their retention times did not overlap with retention times of analytes and peak shapes at each analytical condition. In general, the selection of suitable mobile phases, elution modes, and columns is very important for optimal quantitation of analytes.Thus,different types of mobile phase composition and columns were tested in this study.For simultaneous determination of xanthotoxol, imperatorin, oxypeucedanin,18-beta-glycyrrhetinic acid,glycyrrhizin,atractylenolide III,ferulic acid, methyl eugenol,baicalin,baicalein, atractylenolide I,and notopterol(including repaglinide as an IS),tests were performed with KINETEX core-shell C18column (50 mm × 2.1 mm i.d.,1.7 μm particle size; Phenomenex Inc., CA, USA), HALO-C18column(100 mm × 2.1 mm i.d., 2.7 μm particle size; Advanced Materials Technology Co., DE, USA), and UPLC®BEH C18column(50 mm×2.1 mm i.d.,1.7 μm particle size;Waters Co.,MA,USA).As a result,a better peak sensitivity was observed with the KINETEX coreshell C18column. Acetonitrile and methanol were compared as mobile phase B.Acetonitrile had lower column pressure and better sensitivity of peaks than methanol although there was no significant difference in column pressure or sensitivity between methanol and acetonitrile.Furthermore,when 0.1%(V/V)of formic acid was added to mobile phase A, it restrained peak tailing and peak symmetry. A gradient elution program was applied to completely separate the retention times of 14 ingredients.For simultaneous determination of catalpol and aucubin(including celecoxib as an IS),the Inertsil C8-3 column(100 mm×2.1 mm i.d.,2.0 μm particle size;GL Sciences Inc.,Tokyo, Japan) was selected. The Inertsil C8-3 column was used because iridoid-type materials such as catalpol and aucubin were not highly retained in the C18column,which had relatively high polarity.Therefore,peaks of analytes were not completely separated and the retention time was too short to suspend the void volume. As reported previously[23],acetonitrile and 10 mM aqueous ammonium formate supplemented with 0.01%(V/V)formic acid were optimized as a mobile phase for simultaneous determination of catalpol and aucubin.Buffer salts facilitated stable ionization and retention in the column of catalpol and aucubin. Furthermore, a gradient elution program was used instead of isocratic elution to improve the sensitivity and peak shapes of analytes.As a result,satisfactory separation of xanthotoxol, imperatorin, 18-beta-glycyrrhetinic acid, glycyrrhizin, atractylenolide III, atractylenolide I, ferulic acid, oxypeucedanin,baicalin,methyl eugenol,baicalein,and notopterol was achieved with KINETEX core-shell C18column at a run time of 5 min and a flow rate of 0.3 mL/min.However, catalpol and aucubin were optimized with Inertsil C8-3 column at a run time of 5 min and a flow rate of 0.2 mL/min.Fig.2 presents peaks in LLOQ and ULOQ of each analyte and representative MRM chromatograms.
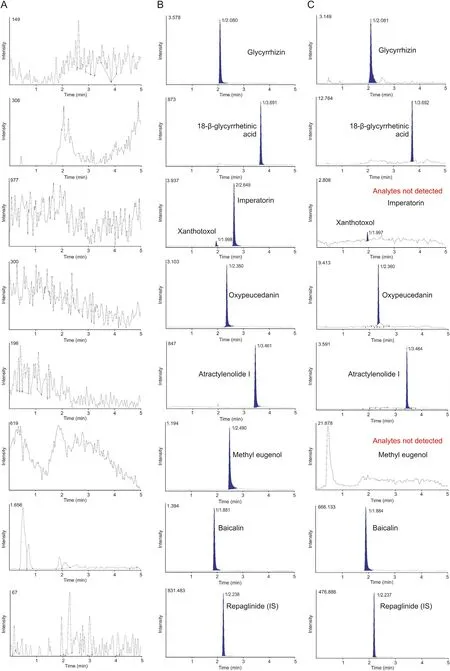
Fig. 3. MRM chromatograms of (A) blank human plasma, (B) blank plasma containing 2 ng/mL of 14 herbal compounds and internal standards (IS), and (C) plasma sample at 2 h after oral administration of Gumiganghwal-tang tablet.

Fig. 3. (continued).

Table 1 Precision and accuracy of UPLC-ESI-MS/MS analysis of 14 compounds in human plasma (mean ± SD, n=5).

Table 2 Recovery and matrix effect of 14 herbal compounds in human plasma(mean±SD,n =5).
To ensure accurate quantification of analytes inbiological samples,sample preparation is very important.Repeated trials are needed to optimize sample preparation.LLE and PP methods provide relatively clean samples at low cost and short time.For LLE,dichloromethane,ethyl acetate,n-hexane,and di-ethyl ether were used for extraction.For the 14 herbal compounds,ethyl acetate supplemented with 1%(V/V)acetic acid extracted the largest amount compared with dichloromethane,di-ethyl ether,or n-hexane,probably because mostof the14 analytes exhibit appropriate polarity and therefore are less compatible with nonpolar solvents(such as dichloromethane,di-ethyl ether,and n-hexane). Also, the PP method using acetonitrile or methanol was tested.Acetonitrile did not yield higher recovery than methanol.Therefore,methanol was selected as the precipitation solvent in the PP method. Above all, methanol yielded a cleaner matrix with less noise than acetonitrile. Therefore, methanol and ethyl acetate supplemented with 1% (V/V) acetic acid were selected and extraction efficiencies of analyteswere compared accordingtotheir proportions.Eventually,the best extraction efficiency was obtained when methanol and ethyl acetate containing 1%(V/V)acetic acid(8:2,V/V)were used in the extraction process.In the present study,the supernatant after extraction was evaporated to dryness under a high purity nitrogen stream at 25°C to improve the sensitivity of 14 components based on a previous study[5].
3.2. Method validation
3.2.1. Selectivity
Selectivity was confirmed by responses to blank plasma, blank plasma spiked with standards and IS and to plasma sample 2 h after oral administration of Gumiganghwal-tang tablets. Representative chromatograms presented in Fig. 3 suggest the lack of significant interference from endogenous substances around the retention times of analytes and IS in the blank plasma.
3.2.2. Calibration curves
Linearities for 18-beta-glycyrrhetinic acid, glycyrrhizin, xanthotoxol, imperatorin, baicalein, baicalin, methyl eugenol, oxypeucedanin, notopterol, atractylenolide III, atractylenolide I, and ferulic acid in human plasma were excellent over concentration ranges of 0.5-500, 0.1-500, 0.5-500, 0.1-500, 0.5-500, 0.5-500,1-500, 0.5-500, 0.1-500, 0.5-500, 0.1-500, and 0.5-500 ng/mL,respectively.Calibration curves showed excellent fit,with correlation coefficient (r2) exceeding 0.99. Linear regression equations were as follows:y =(1.57e-3±1.00e-4)x +(1.44e-4±1.06e-5)for glycyrrhizin,y =(1.02e-4±1.10e-5)x +(1.96e-5±2.25e-6)for18-beta-glycyrrhetinic acid,y =(5.12e-4±1.43e-5)x +(4.24e-5±1.45e-6)for imperatorin,y =(2.03e-4±1.01e-5)x +(1.49e-5±1.12e-6)for xanthotoxol,y =(4.98e-4±2.12e-5)x +(3.97e-5±1.19e-6)for baicalin,y =(1.13e-4±1.10e-5)x +(1.08e-5±1.56e-6)for baicalein,y =(1.41e-3±1.02e-4)x +(1.32e-4±1.28e-5)for notopterol,y =(4.89e-4±1.97e-5)x +(2.50e-5±1.11e-6)for oxypeucedanin, y =(1.99e-4± 1.18e-5)x +(1.48e-5± 2.41e-6) for methyl eugenol,y =(1.90e-4±1.13e-5)x +(1.21e-5±1.04e-6)for ferulic acid,y =(1.01e-4±1.06e-5)x +(1.43e-5±1.05e-6)for atractylenolide I,and y =(5.71e-4±1.44e-4)x +(4.35e-5±1.16e-6)for atractylenolide III in human plasma,where x(ng/mL)denotes the plasma concentration of ingredients and y refers to the peak-area ratio of ingredients to IS.As mentioned above(Section 2.6.2),the dilution integrity(for baicalin)was also determined,and the sample dilution did not affect the accuracy and precision(within±15%).
3.2.3. Accuracy and precision
During the validation, the four QC samples showed robust performance with consistent accuracy and low deviation. The intrabatch accuracy ranged from 97.60% to 105.88% for 14 ingredients,with precision (CV) less than 10.20% in human plasma. The interbatch accuracy ranged from 96.90% to 104.69% for 14 ingredients,with precision (CV) less than 8.33% in human plasma. Table 1 presents inter- and intra-batch accuracy and precision results for the 14 ingredients in human plasma,showing that the accuracy and precision of this quantification method were acceptable.The newly developed method was reproducible and accurate for quantification of 14 ingredients of Gumiganghwal-tang in human plasma.
3.2.4. Matrix effect and recovery
Extraction recoveries of 14 herbal metabolites from human plasma ranged from 75.80% to 96.44%, with IS of 97.31%-99.46%,with no significant matrix effects on their quantification (96.37%-101.14%). In addition, IS (99.09%-100.25%) was the same. Table 2 presents matrix effects and recoveries for 14 components. These results indicated that the analyte recoveries were reproducible,precise, and consistent. Such simple LLE and PP procedures were successfully applied to the determination of 14 herbal compounds of Gumiganghwal-tang in human plasma.

Table 3 Stability of 14 herbal compounds in human plasma under various conditions (mean ± SD, n =5).
3.2.5. Stability
The 14 components were stable in human plasma at 25°C for 24 h without any significant degradation (stability range, 95.39%-102.02%).Inthelong-termstability test afterstorage at-80°C forfour weeks, the 14 components were stable (stability range, 94.51%-102.34%), thus ensuring the quality of quantification after sample collectionwithinfour weeks.In the freeze-thawcycle test,all analytes were stable after three cycles (stability range, 96.73%-102.24%).Furthermore,in autosampler stability,14 compounds were stable at 15°C for 24 h (stability range,95.92%-101.19%). These ranges were within limits of FDA guidelines (±15%) for all stability tests. After assessing the stability of stock solutions of the 14 components and IS,all were stable after storage at-20°C for four weeks.Results of stabilityare presented inTable 3.These results indicate the stability of 14 components under various storage conditions.
3.2.6. Carryover
As shown in Fig. 3A, no clear analyte peaks were observed in blank plasma sample after injecting the highest concentration sample of the standard curve.Thus,there was no carryover effect in this analytical experiment.
3.2.7. ISR
Twenty clinical samples were used to determine ISR.Compared with initially analyzed values, all 20 samples were within 20%variability. Furthermore, variability was within 15% between the mean of reanalysis and that of initial analysis for all samples.Thus,it can be concluded that the developed method shows reproducibility of initial results of analysis.
3.3. PK study
The validated UPLC-ESI-MS/MS method was used in the PK study of 14 ingredients after oral administration of Gumiganghwaltang tablets (9.6 g containing 97403.22 μg of baicalin,10001.25 μg of baicalein, 9573.90 μg of glycyrrhizin, 137.71 μg of 18-beta-glycyrrhetinic acid, 1.30 μg of imperatorin, 26.58 μg of xanthotoxol,10.80 μg of aucubin, 7.61 μg of catalpol, 85.40 μg of ferulic acid,0.70 μg of notopterol, 1235.38 μg of oxypeucedanin, 0.69 μg of methyl eugenol, 31.07 μg of atractylenolide I, and 327.35 μg of atractylenolide III)to 12 healthy Korean subjects.The absorption of most active components was relatively rapid after oral administration. Plasma concentrations close to the Cmaxappeared within 1-2 h after administration. In addition, most of the active ingredients remained in the body for a considerably long time for quantification in plasma 48 h after oral administration.The half-life of oxypeucedanin was 70.55 ± 10.63 h, followed by that of ferulic acid, glycyrrhizin, atractylenolide III, and atractylenolide I. However, the half-lives of 18-beta-glycyrrhetinic acid, baicalin, baicalein,and xanthotoxol were relatively short.The concentration-time curves of 18-beta-glycyrrhetinic acid, glycyrrhizin, baicalin, baicalein, oxypeucedanin, atractylenolide III, atractylenolide I, xanthotoxol, and ferulic acid are presented in Fig. 4. The PK parameters including AUC0-∞, Vd/F, Cmax, t1/2, Tmax, and CL/F are presented in Table 4.The PK parameters determined in this study were difficult to compare with those reported previously because most reported PK parameters were determined in rats(not humans)and/or mixed with other components inducing drug interactions and/or PK results in different formulations (such as micelle and nanoparticle).Therefore, the PK parameters and results obtained from humans with herbal medicines that are widely used clinically are critical to clinical application.
The CL/F and Vd/F of glycyrrhetinic acid, baicalein, and xanthotoxol could not be calculated because they are metabolites of glycyrrhizin,baicalin,and imperatorin,respectively.Since the dosages of only metabolites were not known exactly, the PK parameters of CL/F and Vd/F could not be calculated. In order to accurately calculate the CL/F and Vd/F of glycyrrhetinic acid, baicalein, and xanthotoxol, experiments via oral administration without parent drugs (glycyrrhizin, baicalin, and imperatorin) are necessary.
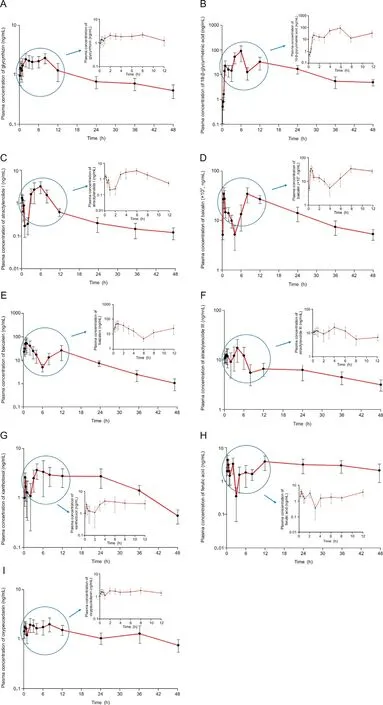
Fig. 4. Mean plasma concentration-time profiles of (A) glycyrrhizin, (B) 18-beta-glycyrrhetinic acid, (C) atractylenolide I, (D) baicalin, (E) baicalein, (F) atractylenolide III, (G) xanthotoxol,(H)ferulic acid,and(I)oxypeucedanin after oral administration of Gumiganghwal-tang tablets(97403.22 μg of baicalin,10001.25 μg of baicalein,9573.90 μg of glycyrrhizin,137.71 μg of 18-beta-glycyrrhetinic acid,1.30 μg of imperatorin,26.58 μg of xanthotoxol,10.80 μg of aucubin,7.61 μg of catalpol,85.40 μg of ferulic acid,0.70 μg of notopterol,1235.38 μg of oxypeucedanin, 0.69 μg of methyl eugenol, 31.07 μg of atractylenolide I, and 327.35 μg of atractylenolide III). Vertical bars represent standard deviation of the mean.

Table 4 Major pharmacokinetic parameters of nine herbal compounds in humans after oral administration of Gumiganghwal-tang tablets(with each 9.6 g containing 97403.22 μg of baicalin,10001.25 μg of baicalein, 9573.90 μg of glycyrrhizin,137.71 μg of 18-beta-glycyrrhetinic acid,1.30 μg of imperatorin, 26.58 μg of xanthotoxol,10.80 μg of aucubin,7.61 μg of catalpol, 85.40 μg of ferulic acid, 0.70 μg of notopterol,1235.38 μg of oxypeucedanin, 0.69 μg of methyl eugenol, 31.07 μg of atractylenolide I, and 327.35 μg of atractylenolide III, mean ± SD).
Concentration-time curves of imperatorin, methyl eugenol,notopterol, aucubin, or catalpol could not be drawn. Their PK parameters could not be calculated because of fewer concentrations detected, which might be related to the composition of Gumiganghwal-tang tablets.Thus,the five compounds(notopterol,imperatorin, methyl eugenol, aucubin, and catalpol), which were not detected in human plasma, were too low (with a mean concentration of less than 1.125 μg/g) in Gumiganghwal-tang tablets based on the composition determined (Section 2.2). As a result,although the Gumiganghwal-tang tablets were orally administered at the maximum daily dose (9.6 g), the PK results of these five components were unfortunately not obtained.
Results showed that baicalein, atractylenolide III, 18-beta-glycyrrhetinic acid, ferulic acid, atractylenolide I, oxypeucedanin,xanthotoxol,glycyrrhizin,and baicalin had double peaks in plasma concentration-time profiles. The appearance of the secondary or multiple peaks may be related to dual absorption or enterohepatic circulation [24]. Glycyrrhizin and baicalin have been reported to enter enterohepatic circulation via bile excretion in rats [25,26].Although atractylenolide III and ferulic acid were not clearly identified in enterohepatic circulation, the PK graphs showed multiple peaks[27,28].Enterohepatic circulation leads to extended residual time of substance in blood and delayed excretion [8].Thus, these findings may be related to our PK results suggesting the presence of active ingredients in human plasma until 48 h after a single oral administration of Gumiganghwal-tang tablets.According to a previous report, many herbal medicines showed multiple peaks in time-plasma concentration graphs as a result of PK studies in vivo, suggesting enterohepatic circulation [8].Additionally, the existence of multiple peaks in the PK graph of many herbal medicines may be attributed to the interaction between various compounds in herbal medicines. In order to elucidate the mechanism of enterohepatic circulation of herbal components, it is necessary to investigate the interaction between phytoconstituents in the future. In addition to enterohepatic circulation, our PK results (double peak phenomena in plasma concentration-time profiles) may be related to dissolution and absorption of the administered formulations. In this PK study, 12 tablets (9.6 g, at the maximum daily oral dose) of Gumiganghwaltang tablets were administered orally and were eventually absorbed in the stomach or intestine. In order to explain the multiple peaks in PK graphs related to dissolution and absorption of the formulation, a comprehensive evaluation of the Gumiganghwal-tang tablet formulation is required.
While additional studies will be conducted in the future,the PK results involving healthy Korean adults in this study are expected to provide useful data for the scientific safety and efficacy of Gumiganghwal-tang tablets, which are still prescribed in clinical practice. Thus, among the PK parameters shown in Table 4, Cmaxand AUC may be related to drug safety or toxicity.In this study,oral administration of Gumiganghwal-tang tablets to humans resulted in significant levels of baicalin in the blood (Cmaxaveraged 34327 ng/mL and AUC0-∞averaged 805216 ng h/mL), which may warrant safety studies (especially at multiple doses). However,when the maximum daily dose of Gumiganghwal-tang was orally administered, nine components were detected in the blood up to 48 h after administration, which may indirectly confirm the effectiveness of Gumiganghwal-tang. Further studies will be needed to correlate blood concentrations with therapeutic effects in order to establish the efficacy. In addition, the t1/2of the PK parameters presented in Table 4 may represent an important basis for the control of the interval of drug administration. In the case of oxypeucedanin,a prolonged t1/2of 70.55 h(as mean)suggests possible accumulation in the body when multiple doses of Gumiganghwaltang are administered at frequent intervals.
4. Conclusion
Based on modification of our method reported previously, the UPLC-ESI-MS/MS method was newly developed and fully validated to simultaneously determine the concentrations of 14 ingredients of Gumiganghwal-tang tablets in human plasma. Our results demonstrate that bioactive compounds of Gumiganghwal-tang can be quantified rapidly, accurately, sensitively, selectively, and relatively simply in biological samples by using the developed UPLCESI-MS/MS in this study.This method was used to characterize PKs of 14 bioactive compounds in humans after oral administration of Gumiganghwal-tang tablets. It was confirmed that oral absorption of the components of Gumiganghwal-tang was relatively rapid and that the active ingredients were quantified in plasma samples up to 48 h after administration. And multiple peak patterns were confirmed in the plasma concentration-time profiles. As a result,this study provides valuable clinical PK data regarding Gumiganghwal-tang tablets, which are often used in clinical practice.The PK profiles of these phytoconstituents will serve as a basis for the design of dosage forms of Gumiganghwal-tang tablets with optimal efficacy and minimal toxicity.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by a grant from the National Development Institute of Korean Medicine (NIKOM) funded by the Korean Ministry of Health and Welfare(MOHW),Republic of Korea.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.08.003.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- A simplified LC-MS/MS method for the quantification of the cardiovascular disease biomarker trimethylamine-N-oxide and its precursors
- UHPLC-MS/MS analysis of cAMP and cGMP in rat plasma as potential biomarkers of Yin-Yang disharmony in traditional Chinese medicine
- Liquid chromatography-mass spectrometry method for the quantification of an anti-sclerostin monoclonal antibody in cynomolgus monkey serum
- Plasma-metabolite-based machine learning is a promising diagnostic approach for esophageal squamous cell carcinoma investigation
- Reducing SARS-CoV-2 pathological protein activity with small molecules
- Development of the general chapters of the Chinese Pharmacopoeia 2020 edition: A review