Modification of total and phosphorus mineralizing bacterial communities associated with Zea mays L.through plant development and fertilization regimes
2021-09-10XlNYuanyuanAnisurRAHMANLlHuixiuXUTingDlNGGuochunLlJi
XlN Yuan-yuan,Anisur RAHMAN,Ll Hui-xiu,XU Ting,,DlNG Guo-chun,,Ll Ji,
1 College of Resources and Environmental Sciences,China Agricultural University,Beijing 100193,P.R.China
2 Organic Recycling Institute (Suzhou),China Agricultural University,Suzhou 215128,P.R.China
Abstract Harnessing the rhizospheric microbiome,including phosphorus mineralizing bacteria (PMB),is a promising technique for maintaining sustainability and productivity in intensive agricultural systems.However,it is unclear as to which beneficial taxonomic group populations in the rhizosphere are potentially associated with the changes in soil microbiomes shifted by fertilization regimes.Herein,we analyzed the diversity and community structure of total bacteria and PMB in the rhizosphere of maize (Zea mays L.) grown in soils under 25 years of four fertilization regimes (compost,biocompost,chemical,or nonfertilized) via selective culture and Illumina sequencing of the 16S rRNA genes.Plant development explained more variations(29 and 13%,respectively) in the composition of total bacteria and PMB in the rhizosphere of maize than the different fertilization regimes.Among those genera enriched in the rhizosphere of maize,the relative abundances of Oceanobacillus,Bacillus, Achromobacter, Ensifer,Paracoccus,Ramlibacter,and Luteimonas were positively correlated with those in the bulk soil.The relative abundance of Paracoccus was significantly higher in soils fertilized by compost or biocompost than the other soils.Similar results were also observed for PMB affiliated with Ensifer, Bacillus, and Streptomyces.Although plant development was the major factor in shaping the rhizospheric microbiome of maize,fertilization regimes might have modified beneficial rhizospheric microbial taxa such as Bacillus and Ensifer.
Keywords:organic fertilization,bacterial diversity,phosphorus mineralizing bacteria (PMB), Zea mays L., rhizosphere
1.lntroduction
Phosphorus (P) is one of the major nutrients required by crops and it is essential for agriculture;however,reserves of high-quality phosphate rock are limited (Mogollónet al.2018).Presently,high crop productivities often depend on external nutrient inputs.The excessive use of P is problematic in several aspects,such as soil P-surplus (Wanget al.2015),water eutrophication (Hashemiet al.2016),and biodiversity loss (Hautieret al.2009).Soluble P can be immobilized soon after field fertilization,and P-surplus in arable soils is common in the North China Plain (Xinet al.2019).Solubilizing P in soils for crops is a potential strategy for simultaneously maintaining relatively high crop productivity and mitigating agricultural-source pollution.Members of microbial genera such asPseudomonas,Bacillus,andEnterobacteriacan harness phytases,acid,or alkaline phosphomonoesters (Aloriet al.2017;Sharmaet al.2013),to mineralize organic P,which accounts for up to 80%of the soil P pool (Sharmaet al.2013).The application of P solubilizing/mineralizing microbes can enhance the growth of several crops such as winter wheat (Kämpferet al.2005),rice (Souzaet al.2015),sorghum (Rezakhaniet al.2019),maize (Babalolaet al.2003;Bhattacharyya and Jha 2012;Ogbo and Okonkwo 2012),tomato (Elthbanyet al.2019),and Chinese cabbage (Wanget al.2017).However,the effects of P mineralizing/solubilizing microbial inoculants have often proven to be variable,especially under field conditions (Aloriet al.2017;Sharmaet al.2013).A better understanding of the ecology between P mineralizing bacteria (PMB) and crops is needed to harness PMB as a tool for promoting plant growth.
Agricultural management may influence the abundance,diversity,and activity of microorganisms associated with P cycling in soil (Daiet al.2020;Spohnet al.2015).For example,low-P fertilization could possibly enhance microbial competence for P in soil,as suggested by the increased acid or alkaline phosphatase activities (Spohnet al.2015)or the relative abundance of populations containing the P-starvation response gene (phoR) (Daiet al.2020).Long-term organic fertilization could also result in several changes in soil,including shifts in the microbial functional communities associated with P-or N-cycling (Dinget al.2019) and taxonomic composition (Hartmannet al.2015),soil carbon,and nutrient element contents (Gattingeret al.2012).Soil serves as a seed bank which allows plants to recruit their rhizospheric microbiomes.Changes in soil may also influence the assembly of the rhizospheric microbiomes of several crops such as rice (Tanget al.2019),tomato(Allardet al.2016),soybean (Zhanget al.2020),and maize(Zhanget al.2020).The underlying mechanisms governing the bacterial assembly are complicated,and several abiotic and biotic factors may be involved (Fierer 2017).It is possible that changes in specific soil microbial taxa may also influence their abundance in the rhizosphere.However,knowledge on rhizospheric taxa that can be potentially modifiableviaagricultural management is insufficient.
Herein,we used a 25-year field experiment with different fertilization regimes to study the diversity,community composition,and succession of total bacteria and PMB in the bulk soil and rhizosphere of maize (Zea maysL.) at four developmental stages of its lifecycle.The specific aims of this study are:1) to decipher the effects of different longterm fertilization regimes on the diversity and community composition of total bacteria and PMB in bulk soils and the rhizosphere of maize;2) to identify key rhizospheric taxa and PMB that might be modified by different fertilization regimes.
2.Materials and methods
2.1.Experimental set-up and sampling
The long-term field experiment was initiated in 1993 at Quzhou Experimental Station (36° 52´N,115° 01´E),Hebei Province,China.The fertilization regimes used in this study included compost (15 t ha-1yr-1) or biocompost (15 t ha-1yr-1),chemical fertilizer (N-P2O5-K2O at 200-250-270 kg ha-1yr-1),and non-fertilized,using a randomized block design with four replicates.In the chemically fertilized soil,urea,superphosphate,and potassium sulfate were used as the nitrogen,phosphorus,and potassium fertilizer.Each replicate covered an area of 31.5 m2(10.5 m×3 m).Winter wheat (Triticum aestivumL.) and summer maize (Zea maysL.) were rotated each year.Details on the characteristics of the bulk soil are provided in Table 1.Bulk soil and rhizospheric samples were taken at the seedling (10 July),jointing (23 July),trumpet mouth (4 August),and tasseling(16 August) stages of maize in 2018.Each soil sample consisted of five cores (each 2 cm in diameter) of soil,at a depth of 1-20 cm.All soils were passed through a 2-mm mesh to remove stones and plant debris.Each rhizospheric sample consisted of the root systems from three individual plants,and all samples were transferred to the lab using a cooling box (ca.4°C).Rhizospheric samples were obtained by 10 periods of vigorous shaking in 0.85% NaCl solution,to remove the adhering soil (Barillotet al.2013).A total of 1 mL of this mixture was used for the isolation of the PMB.The remaining rhizospheric samples were centrifuged at 3 000×g for 5 min to collect pellets for DNA extraction.All samples were kept at -20°C prior to DNA extraction.
2.2.Soil physicochemical properties
Bulk soil physicochemical parameters such as pH,moisture,bulk density,Olsen phosphorus,available potassium,organic matter,and total nitrogen were analyzed according to standard protocols (Bao 2000).The pH was measured with a glass electrode in a 1:5 ratio soil/water solution.The moisture content was measured by drying at 105°C for 24 h.The bulk density of soil was measured using a cylinder of known volume containing an undisturbed soil sample.Olsen phosphorus was measuredviathe molybdate blue colorimetric method.The available potassium was determined using flame emission spectrometry.Total nitrogen was determined using the method of Kjeldahl.Soil organic matter was determined using the potassium dichromate oxidation method.

2.3.Selective culture,BOX-PCR,and 16S rRNA sequencing of PMB
Both bulk soil and rhizospheric samples were subjected to serial dilution using 0.85% NaCl solution.PMB were isolated using the methods described by Zhouet al.(2011),using an organic phosphorus medium (glucose 10 g;CaCO35 g;NaCl 0.3 g;MgSO4·7H2O 0.3 g;KCl 0.3 g;MnSO4·4H2O 0.03 g;FeSO4·7H2O 0.03 g;egg yolk 1.0 g;(NH4)2SO40.5 g;and agar 16 g for 1 L) after incubation at 30°C for three to seven days.Genomic DNA extraction,BOX-PCR,PCR amplification of 16S rRNA,and the following Sanger sequencing techniques were mainly performed according to the methods of Liet al.(2019a).The genomic DNA of each isolate was extracted using the bacterial genomic DNA Extraction Kit (Beijing Biomed Co.,Ltd.,China).BOX-PCR was performed according to the methods of Adesinaet al.(2007),using BOX-A1R primers (5´-CTACGGCAAGGCGACGCTGACG-3´) (Louwset al.1994).The PCR reaction mixture was prepared as follows:6.4 µL of ddH2O;1.6 µL of BSA;2.5 µL of DMSO;12.5 µL of TaKaRa mix (containing Mg2+,dNTPs,Taq;TaKaRa Biomedical Technology Co.,Ltd.,China);1 µL of BOX-A1R primers;and 1 µL of template DNA,in a final volume of 25 µL.Thermocycles comprised 7 min at 94°C and 35 cycles consisting of 1 min at 94°C,1 min at 53°C,and 8 min at 65°C.A final extension at 65°C for 16 min was added.The BOX-PCR products were analyzed by electrophoresis in 1.0% agarose gel.The molecular fingerprints were analyzed using the Software GelJ (Heraset al.2015).The fingerprinting profiles were aligned according to the bands of 5 kb DNA ladder,the profile was franked,and then metric curves were extracted after de-noising.The unweighted pair-group method with arithmetic mean clustering was performed,based on the pairwise Pearson correlation coefficient between the curve metrics.PMB were assigned to different BOX-PCR groups when their profile similarity was less than 80% (Heueret al.2007).The 16S rRNA gene fragments of representative isolates of each BOX-PCR pattern were amplified with 27F (5´-AGAGTTTGATCATGGCTCAG-3´)and 1492R (5´-TACGGTTACCTGTTACGACTT-3´) (Suzukiet al.1996),and the PCR product was gel-purified prior to bidirectional Sanger sequencing.Only the regions franked by 515F(5´-GTGCCAGCMGCCGCGGTAA-3´) and 909R (5´-CCCCGYCAATTCMTTTRAGT-3´)(Caporasoet al.2010) were truncated.Unique 16S rRNA gene fragments regarded as a phylotype of PMB were retrieved using the Software Package VSEARCH (Rogneset al.2016)to estimate the composition of potential PMB in the rhizosphere and bulk soil.
2.4.High throughput sequencing analysis of bacterial 16S rRNA gene amplicons
Total community DNA was extracted using a FastDNA Spin Kit for Soil (MP,Biomedicals,Santa Ana,Carlsbad,CA,USA) according to the manufacturer’s instructions.The amplification of fragments of the 16S rRNA gene was performed with the universal primers 515F and 909R,with a 12-nt unique barcode for each sample (Caporasoet al.2010).Gel-purification of the 16S rRNA gene amplicon was performed according to the previous description (Dinget al.2019).Sequencing was performed with the Illumina Hiseq2500 Platform.Only sequences with high quality (length>300 bp,without ambiguous base“N”and average base quality score>30)were used for downstream analyses.Sequences were assigned to each sample based on barcodes,and technical regions (primer and barcode) were trimmed out for the following analysis.Chimera sequences were removed jointly by a standalone BLASTN analysis based on a SILVA bacterial database V132 (Pruesseet al.2007),and a ChimeraSlayer analysis(Haaset al.2011).Operational taxonomic unit (OTU) (>97% sequence identity) assignment and classification were performed with Software QIIME 1.9 and RDP MultiClassifier using the“trainset 16”,respectively (Caporasoet al.2010).Principal coordinates analysis (PCoA) was performed using the Bray-Curtis distance,based on the relative abundances of different genera.Discriminative taxa among fertilization regimes or between bulk soil and rhizosphere were identified by multiple comparisons under a negative binomial model using the R add-on package“multcomp”(Hothornet al.2017).Discriminative genera were re-organized by cluster analysis,based on a pairwise distance that measures the dissimilarity between the responses of different genera to the fertilization regime or rhizosphere.For those genera enriched in the rhizosphere,Spearman and Pearson correlation coefficients were calculated between their relative abundances in the rhizosphere and the bulk soil.A standalone BLASTN analysis was performed to identify the 16S rRNA genes that shared high similarity (>99%identity) with those of PMB from the amplicon sequencing library by the Illumina Hiseq2500 Platform.A data frame was created with each row representing a phylotype of PMB containing taxonomic information (domain,phylum,class,order,family,and genus) and the relative abundance for each sample.Alpha diversity indices (Chao1 and Shannon)were also performed using 100 repeats of resampling of the same amount of reads for each sample (Dinget al.2012).This method allows us to reduce the bias caused by the different numbers of sequence reads per sample.All statistical analyses and the plot were performed with the Software R 3.3 (http://www.r-project.org/),and these tools were implemented into the galaxy instance (www.freebioinfo.org).All sequences were submitted to NCBI SRA (PRJNA578227).
3.Results
3.1.Fertilization regime shifted bacterial community more in the bulk soil than in the rhizosphere of maize
Both rhizospheric samples and bulk soils were analyzed at four growth stages of maize;a total of 4 053 293 reads were acquired for 128 samples from the Illumina sequencing of 16S rRNA fragments.As expected,most sequences were affiliated with Proteobacteria,Actinobacteria,Acidobacteria,Bacteroidetes,and Firmicutes (Appendix A).PCoA analysis revealed that the bacterial community compositions were drastically different between the rhizosphere and the bulk soil (Fig.1-A).Variability in beta-diversity was higher in the rhizosphere (50.5% dissimilarity) than in the bulk soil(33.8% dissimilarity) (Fig.1-A).Variation partition analysis confirmed that the maize rhizosphere and the sampling time could explain 29 and 13% of the variation in the total bacterial community,respectively.The fertilization regime only explained 3% of variation.For the bulk soil bacterial community,the sampling time and the fertilization regime explained 43 and 20% of the variation,respectively.Bacterial communities at the stage of jointing were different from other sampling times (Fig.1-B).Organic fertilization also caused changes in soil bacterial communities at both jointing and other sampling times (Fig.1-B).In the rhizosphere,the development stage was the major factor in shaping bacterial communities (Fig.1-C),explaining 28% of the observed variation.Bacterial communities at seedling and jointing substantially differed from those at trumpet mouth and tasseling (Fig.1-C).No evident effects of the fertilization regime were observed on the alpha diversity of the bacterial community in either the bulk soil or the rhizosphere of maize (Appendix B).
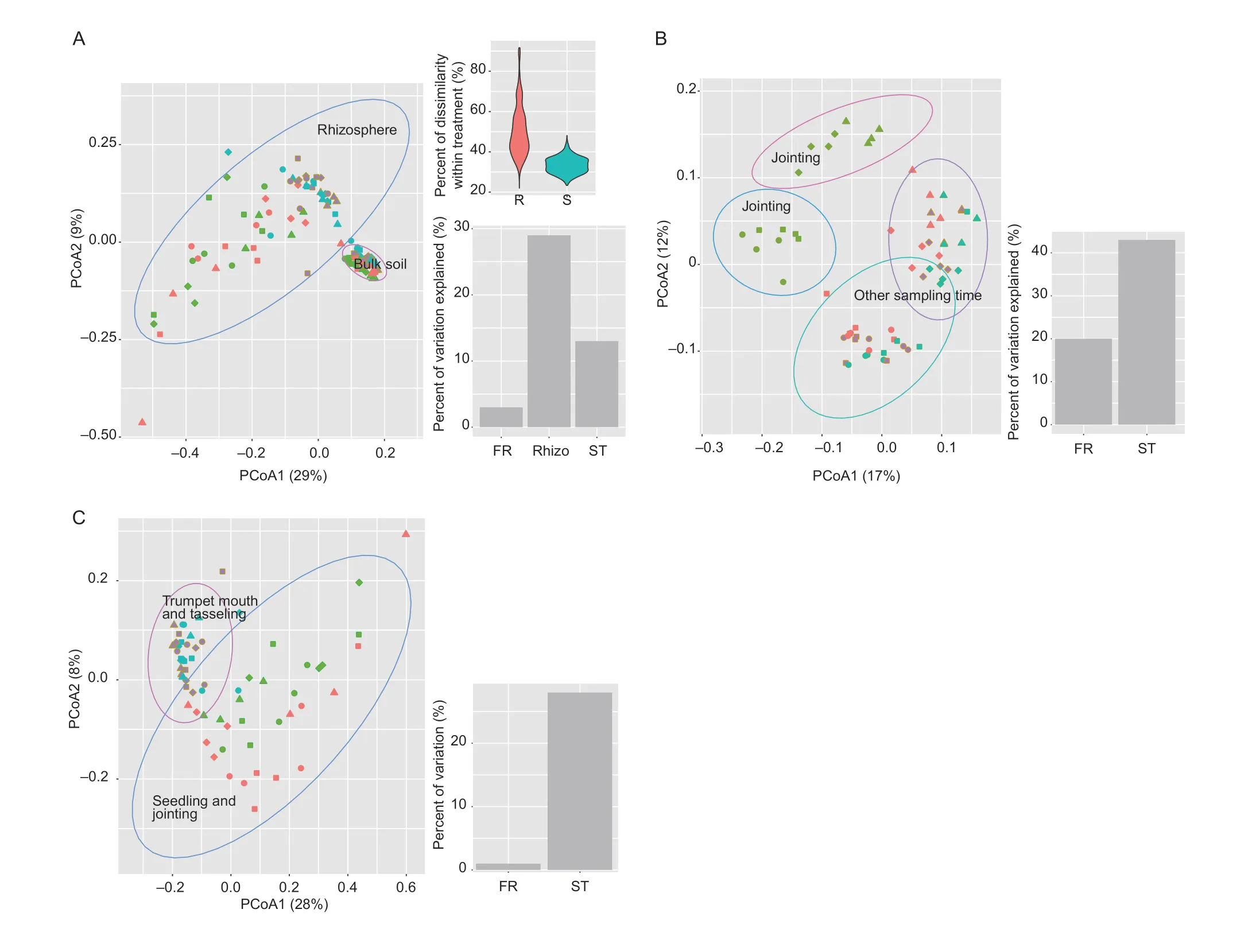
Fig.1 Principal coordinates analysis (PCoA) of the bacterial community under compost (Com),biocompost(Bio),chemical fertilizer (Che),and non-fertilized (Non)treatments,at four sampling times (seedling,jointing,trumpet mouth,and tasseling).A,bulk soil,and rhizosphere.B,bulk soil.C,rhizosphere.Different fertilization regimes are indicated by shapes (circles for biocompost,squares for compost,diamonds for chemical fertilization,and up-triangles for non-fertilized control) and sampling time is indicated by colors (orange for seedling,green for jointing,purple for trumpet mouth and cyan for tasseling).Bar plots indicate percent of variation explained by rhizosphere (Rhizo),sampling time (ST),and fertilization regime (FR) in the bulk soil and rhizosphere.
Discriminative genera among soils under different fertilization regimes were identified and grouped according to their response patterns.Basically,these discriminative genera could be grouped into three response patterns.The first group includedNitrosospiraandNitrosomonas,which tended to be higher in the bulk soil that was chemically fertilized than in the other treatments (Fig.2).The second group included AcidobacteriaGp6,Gp25,Armatimonadetes_gp4,Geminicoccus,Longilinea,andFilomicrobium,which were often lower in organicallyfertilized soils than chemically-or non-fertilized soils (Fig.2).The third group included several genera affiliated with Firmicutes (Oceanbacillus,Gracilibacillus,Ornithinibacillus,Paenibacillus,andTerribacillus),Proteobacteria (Ensifer,Novosphingobium,Microvirga,Achromobacter,andLuteimonas),and Actinobacteria (Actinocorallia,Cellulosimicrobium,Rhodococcus,Saccharomonospora,andNonomuraea),which tended to be higher in the organically-fertilized soils than other treatments (Fig.2).
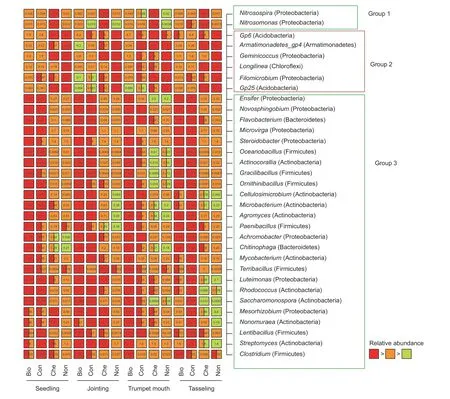
Fig.2 Genera with significantly different relative abundances in the bulk soil under compost (Com),biocompost (Bio),chemical fertilizer (Che) and non-fertilized (Non) treatments,at four sampling times (seedling,jointing,trumpet mouth,and tasseling).Numbers on the boxes indicate relative abundance,expressed as a percentage.Significant differences are indicated by a different colors.A box with two colors indicates no significant difference from the other treatment containing one of the two colors.
3.2.Genera responded to plant development stage and fertilization regime in the rhizosphere of maize
Genera significantly (P<0.05) enriched in the rhizosphere were identified.Interestingly,the effects of the maize rhizosphere on different genera were varied in frequency and magnitude (Fig.3).Persistent,stage-dependent,and occasional response patterns were identified(Fig.3-A).Rhizobium,Ensifer,Sphingobium,Rhizobacter,Shinella,Bacillus,andArthrobacterwere persistently enriched in the rhizosphere (Fig.3-A).Genera affiliated with Betaproteobacteria (Aquabacterium,Acidovorax,Duganella,Noviherbaspirillum,Pelomonas,Ramlibacter,andMassilia),Alphaproteobacteria(Phyllobacterium,Paracoccus,Porphyrobacter,andSphingomonas),Gammaproteobacteria(ButtiauxellaandEnterobacter),Paenibacillus,andLuteolibacterwere mainly enriched at the trumpet mouth and tasseling stages (Fig.3-A).Enterobacter,Buttiauxella,Pantoea,Pseudomonas,Sphingobium,andChryseobacteriumwere occasionally,but strongly,enriched in the rhizosphere (on average>seven-fold) (Fig.3-B).Relative stable enrichment was observed forPaenibacillus,Ensifer,Arthrobacter,andBacillus,suggesting that these rhizospheric genera were less affected by the fertilization regime or the plant development stage (Fig.3-B).
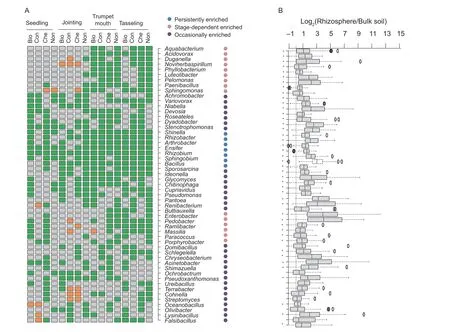
Fig.3 Genera enriched in the rhizosphere at different frequencies (A) and magnitudes (B).Green and orange boxes indicate genera with significantly (P<0.05) higher and lower relative abundance in the rhizosphere than in the bulk soil,respectively.A gray box indicates no significance between the rhizosphere and bulk soil.* indicates P<0.05.
To study whether the abundance of soil bacterial genera may influence their abundance in the rhizosphere,Spearman and Pearson correlation coefficients between their relative abundances in bulk soil and rhizosphere were calculated for those genera enriched in the rhizosphere.Significant positive correlations (P<0.05) were detected forOceanobacillus,Bacillus,Achromobacter,Paracoccus,Ensifer,Ramlibacter,andLuteimonas(Table 2).Interestingly,all these genera(except forParacoccus) were also significantly higher in the organically-fertilized soils than those that were chemically-or non-fertilized (Fig.2),suggesting that organic fertilization might have enriched these genera in soil and consequently increased their abundance in the rhizosphere (Fig.2).
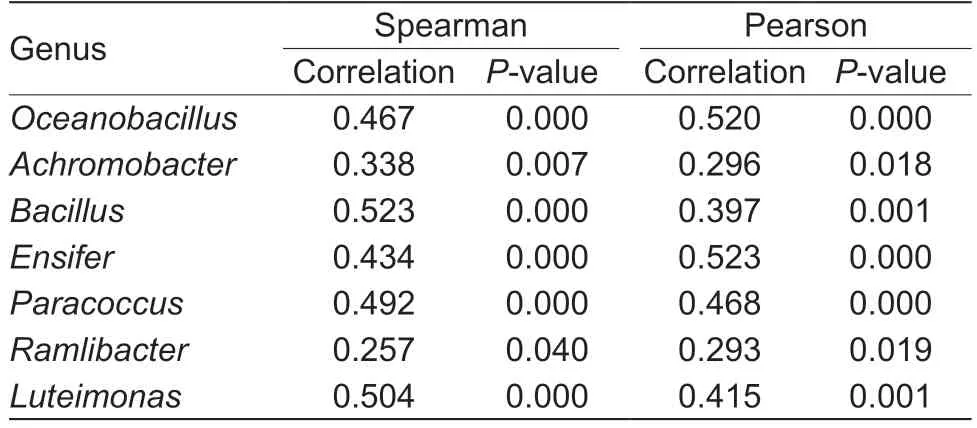
Table 2 Correlations between rhizosphere and bulk soils for genera enriched in the rhizosphere of maize
3.3.Rhizosphere,plant developmental stage,and fertilization regime greatly influenced diversity,abundance,and community composition of PMB
In this study,PMB were isolated by selective culture from both rhizosphere and bulk soils,under different fertilization regimes,at four developmental stages of maize.A total of 1 879 PMB were acquired.BOX-PCR analysis revealed that these PMBs could be grouped into 405 different BOXPCR patterns.Representative isolates of each BOX-PCR pattern were classified into 35 genera,and the majority of PMBs were affiliated withEnterobacter,Stenotrophomonas,Pantoea,Citrobacter,Sphingomonas,Pseudomonas,Bacillus,Rhizobium,Variovorax,andAcinetobacter(Fig.4-A).The relative abundance of PMB affiliated withEnterobacterwas higher in the rhizosphere than in the bulk soil (Fig.4-A).The taxonomic compositions of PMB were significantly different (permutation test:P=0.002,community dissimilarity=10.6%) between the bulk soil and the rhizosphere (Fig.4-B).The majority of these PMB only demonstrated weak (59.2-75.8%) or medium(21.4-38.8%)in vivophosphorus mineralizing activities(Fig.4-C).The fractions of PMB with differentin vivophosphorus mineralizing activities were comparable either among fertilization regimes,or between the rhizosphere and the bulk soil (Fig.4-C).
We additionally integrated Illumina sequencing and selective cultures to identify potential PMB in the bulk soil and rhizosphere of maize.The 16S rRNA gene sequences of 405 representative PMB isolates were grouped into 72unique phylotypes (100% sequence identity).A total of 663 897 16S rRNA gene sequences could be mapped (>99%sequence identity) with these 72 phylotypes,accounting for 16.4% of the total sequences acquired by the Illumina sequencing.The percentages of mapped sequences were higher in the rhizosphere (5.29-78.72%) than in the bulk soil (2.65-6.72%) (Fig.4-D).Regarding the rhizosphere samples,the percentages of mapped sequences were higher at the seedling (34.2+18.1%) and jointing (37.8+14.1%)stages than in the trumpet mouth (14.4+7.8%) and tasseling(11.6+2.9%) stages (Fig.4-D).The majority of mapped sequences were affiliated withArthrobacter,Pseudomonas,Enterobacter,Buttiauxella,Klebsiella,Streptomyces,Citrobacter,Ensifer,Bacillus,andPantoea(Fig.4-D).The rhizosphere rather than the fertilization regime was the major factor influencing the compositions of the potential PMB(Fig.4-E).Twenty-five genera were significantly enriched in the rhizosphere (Appendix C).Among them,Klebsiella,Citrobacter,Buttiauxella,Enterobacter,andPseudomonaswere enriched strongly (on average>seven-fold) in the rhizosphere of maize,followed byArthrobacter,Ensifer,Bacillus,andStreptomyces(Fig.4-F).Significant positive correlations (P<0.05) between their relative abundances in bulk soil and the rhizosphere were observed forEnsifer(Spearman correlation 0.425),Bacillus(Spearman correlation 0.364),andStreptomyces(Spearman correlation 0.343).
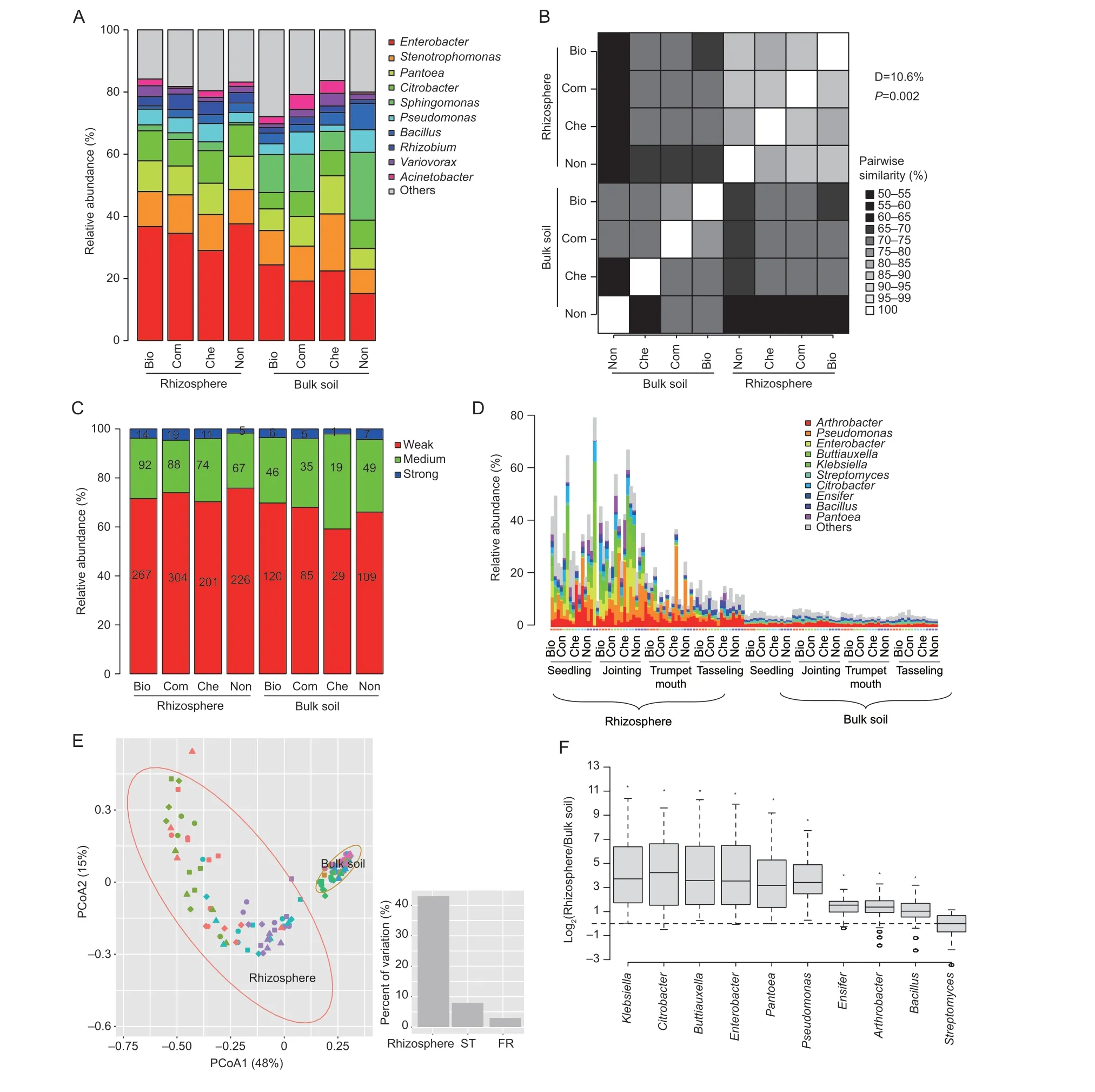
Fig.4 Diversity of phosphorus mineralizing bacteria (PMB) in the bulk soil and rhizosphere under compost (Com),biocompost (Bio),chemical fertilizer (Che),and non-fertilized (Non),by selective culture (A,B and C) and retrieved PMBs from metagenomics (D,E and F).Beta-diversity between samples with similarity is indicated by a gradient of gray colors and the D-values indicate percent of dissimilarity between PMBs composition isolated from rhizosphere and bulk soil (B).In vivo phosphorus mineralizing ability is indicated by the ratio of halo diameter divided by colony diameter to colony diameter (weak,1-2;medium,2-3;strong,>3) and the numbers on the bars indicate the amount of PMB isolated (C).Relative abundance (B),beta diversity (E) and rhizospheric genera enriched at high magnitude (F) of the retrieved PMB.Bar plots indicate percent of variation in the compositions of retrieved PMBs explained by rhizosphere,sampling time (ST),and fertilization regime (FR) using variation partition analysis (E).
4.Discussion
4.1.Long-term organic fertilization shifted microbial communities in the bulk soil
The effects of long-term organic fertilization on soil physiochemical properties observed herein,such as increasing soil organic matter and nutrient (N and P)contents,are in accordance with other studies (Gattingeret al.2012;Hanet al.2019).The increased relative abundances ofNitrosospiraandNitrosomonas,known as ammonia oxidizing bacteria,have also been observed in chemically-fertilized soils (Dinget al.2019).This finding agrees with the finding that ammonia fertilization tends to select for ammonia oxidizing microorganisms.Interestingly,the relative abundance ofLongilineawas also lower in the organically-fertilized soil,and this genus has been found to be prevalent in anaerobic niches,such as anaerobic sludge(Shuet al.2015;Yamadaet al.2007) and methanogenic consortia (Yamadaet al.2007).Thus,organic fertilization possibly selects against strict anaerobic populations despite its introduction of a large amount of organic material into soil which may improve soil porosity (Zhanget al.2015).
Long-term organic fertilization improves soil porosity(Zhouet al.2013;Naveedet al.2014),thereby enhancing oxygen diffusion.However,the effect of organic fertilization on AcidobacteriaGp6contrasted with the findings of a greenhouse experiment conducted at the same experimental station (Dinget al.2019).The experimental conditions,such as crops and field conditions,were drastically different in these two experiments.The long-term growth of different plants may also result in substantial changes in soil microbial communities (Wielandet al.2001;Marschneret al.2004;Lambet al.2011;Dinget al.2013).Thus,the effects of long-term organic fertilization on soil microbial taxa are likely to be context-dependent and to be jointly shaped by several other factors,such as crop rotation and field conditions.
4.2.Maize developmental stage,rather than fertilization regime,was the major driver shaping the rhizospheric microbiome
The physicochemical properties of the rhizosphere are synergistically shaped by plant growth and soil biogeochemical properties,and these characteristics may influence the composition of microorganisms inhabiting the rhizosphere (de la Fuente Cantóet al.2020).Herein,the developmental stage of maize,rather than the fertilization regime,was the major factor shaping the composition of the rhizospheric microbiome.This finding supports the theory that rhizosphere effects are much stronger than soil biotic and edaphic properties regarding the assembly of the microbial community (Chaparroet al.2014).Shifts in microbial communities during plant development have also been reported for other crops such as Arabidopsis (Chaparroet al.2014),maize (Baudoinet al.2002),soybean (Xuet al.2009),and eggplant (Liet al.2019b).The responses of different genera to the maize rhizosphere were varied in frequency and magnitude.Genera includingRhizobium,Ensifer,Sphingobium,Rhizobacter,Bacillus,Shinella,andArthrobacterwere found most frequently enriched in the rhizosphere of maize,suggesting that these taxa were possibly less sensitive to maize development than other taxa.Members ofSphingobium,Bacillus,andArthrobacterare able to utilize a broad spectrum of carbon sources,including lignin and aromatic hydrocarbons (Ahmadet al.2009;Bugget al.2011;Haritashet al.2009).Members ofRhizobium,Ensifer,andRhizobacter,known as nitrogen fixing bacteria,are able to form symbioses with legume plants (Cocking 2003;Francheet al.2009;Masson-Boivinet al.2009;Velázquezet al.2010),and they are also prevalent in non-legume plants such asParasponia andersonii(Santiet al.2013) or eggplant (Liet al.2019b).Enterobacter,Buttiauxella,Pantoea,Pseudomonas,Sphingobium,andChryseobacteriumincreased by great amounts,suggesting that the members of these genera were competent in the maize rhizosphere.Members of these genera have frequently been associated with other crops such as cucumber (Khabbazet al.2015),maize (Peifferet al.2013),wheat (Yinet al.2013),and pepper (Yanget al.2012;Liet al.2019a).
4.3.Long-term organic fertilization elevated beneficial taxa in the rhizosphere of maize,with implications for engineering rhizospheric microbiomes via agricultural management
Engineering the rhizospheric microbiomeviaplant breeding and agricultural management has been raised as a promising strategy for sustainable agriculture (Bakkeret al.2012).Agricultural management techniques such as organic fertilization have likely contributed to the suppression of several soil-borne diseases (Qiuet al.2012;Zhanget al.2017).
Herein,the relative abundances ofOceanobacillus,Bacillus,Achromobacter,Ensifer,Ramlibacter,andLuteimonaswere enriched in both the organically-fertilized bulk soil and the rhizosphere of maize (Figs.2 and 3-A).Interestingly,their abundances in the bulk soil and the rhizosphere were also positively correlated (Table 2),indicating that these microbial taxa might be enhanced in the rhizosphere of maize by increasing their abundance in the bulk soil.Among these genera,members ofBacillus(Fanet al.2018;Liet al.2019a),Oceanobacillus(Masmoudiet al.2019),Achromobacter(Maet al.2009),andEnsifer(Quéréet al.2017) are often reported as plant growth promoting bacteria,or as bioagents against plant diseases.Organic fertilization,among other agricultural management techniques,is likely to contribute to the suppression of several soil-borne diseases (Qiuet al.2012;Zhanget al.2017).Additionally,rhizocompetence,which is the ability to colonize the rhizosphere of a crop at a relatively high density,plays an important role on the performance of the microbial inoculant (Berget al.2017;Elthbanyet al.2019).Members of these genera could be used for engineering the microbiome in the rhizosphere of maize to improve its health and productivity.
4.4.Maize recruited PMB at the seedling and jointing stages
Integrating selective cultures and the Illumina sequencing of 16S rRNA fragments revealed that the relative abundances of PMB were much higher in the rhizosphere than in the bulk soil,suggesting that plants may recruit beneficial bacteria for P-acquisition,even though plants could secrete phosphatase or carbohydrates by themselves.P-solubilizing bacteria have been shown to be enriched in the rhizosphere of eggplant at the flowering and fruiting stages (Liet al.2019b).Herein,the fraction of PMB in the rhizosphere of maize was higher in the early stages than in the late stages.These results seem to be contradictory.Eggplant takes up large volumes of P during the flowering and fruiting stages (Arnonet al.2004),whereas maize needs more P during the early stages (Gavitoet al.1998;Plénetet al.2000).These results highlight that the nutrient demands of plants might be one of the driving forces shaping rhizospheric bacterial communities and for the selection of functional microorganisms.However,notably,only a fraction of PMB may be cultivated in the bulk soil and in the rhizosphere of maize.Further analyses at the single cell level,or of functional genes (phoD;Wanget al.2019),may deepen our understanding of the complicated interactions between microorganisms and crops.
5.Conclusion
In summary,differences in the long-term fertilization regime greatly influenced the soil microbiome.Furthermore,the assembly of the microbiome and the recruitment of PMB in the rhizosphere of maize were more susceptible to plant development than to the fertilization regime.However,beneficial microbial taxa,includingOceanobacillus,Bacillus,Achromobacter,Ensifer,Ramlibacter,andLuteimonas,were enriched correspondingly in bulk soil and the rhizosphere by long-term organic fertilization,highlighting their potential for use in engineering the rhizospheric microbiome of maize.
Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFD1002000,2016YFD0800602 and 2016YFD0501404).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- Errata regarding previously published articles
- Viricidal activity of several disinfectants against African swine fever virus
- Application of methyl jasmonate postharvest maintains the quality of Nanguo pears by regulating mitochondrial energy metabolism
- Melatonin treatment induces chilling tolerance by regulating the contents of polyamine,γ-aminobutyric acid,and proline in cucumber fruit
- Linking changes in the soil microbial community to C and N dynamics during crop residue decomposition
- Yield performance and optimal nitrogen and phosphorus application rates in wheat and faba bean intercropping