生物信息学方法预测病原体抗原蛋白序列中多肽疫苗候选表位
2021-09-08赵静静澹小秀王广志欧阳俭简星星
赵静静,澹小秀,王广志,欧阳俭,简星星,3*,谢 鹭*
(1.上海海洋大学食品学院,中国上海 201306;2.上海市生物医药技术研究院上海生物信息技术研究中心,中国上海 201203;3.中南大学湘雅医院生物信息中心,中国湖南 长沙 410008)
疫苗是当前人类对抗病原生物最经济有效的方式。现阶段,针对病原体蛋白中能够引起免疫系统效应功能的表位而设计的多肽疫苗是新型疫苗研发的热点。多肽疫苗一般是指利用化学合成技术,按照病原体抗原基因中具有免疫原性的一段或多段氨基酸序列构成而制备的疫苗[1]。其因成分简单、特异性强、安全性高、易于保藏等优点被广泛应用于传染病和肿瘤的预防和治疗中,例如:预防人乳头瘤状病毒的疫苗[2]、预防乙型肝炎病毒的HBsAg/preS疫苗[3]、新型艾滋病疫苗RV144[4]及个性化肿瘤新抗原疫苗[5]等。
近年来,得益于生物信息学技术的迅猛发展,反向疫苗学在疫苗设计领域显示出广阔的应用空间。以“序列-结构-功能”思想为依据,借助生物信息学工具对病原体高通量的组学信息(基因组学、转录组学、蛋白质组学等)预先进行严格的预测和筛选,能极大地提高研究人员发现病原体蛋白质序列中候选表位的效率。随着候选表位的持续产出和收集,相关表位数据库应运而生,如IEDB(Immune Epitope Database)[6]及本课题组自行构建的dbPepNeo[7]等。研究人员利用这些数据集开发了诸多表位预测和筛选的工具,但其中的多数均只针对特定的功能进行开发,如:NetMHC-pan EL 4.0[8]主要用于预测与主要组织相容性复合体(major histocompatibility complex,MHC)结合的抗原表位;本课题组开发的INeo-Epp[9]则用于预测具有免疫原性的抗原表位。因此,表位预测工具功能的多样化,对于候选表位的鉴定以及多肽疫苗的设计具有重要指导意义[10~12]。
本研究基于反向疫苗学策略,综合利用多种生物信息学工具,构建了一个完整的、易于操作的表位预测和筛选流程,并以当下正在全球蔓延的严重急性呼吸综合征冠状病毒2(severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)为实例,成功地检验了该流程的实用性。文中共筛选到34条关于SARS-CoV-2的T细胞候选表位,其中20条候选表位与数据库中经过验证的表位高度同源,且能够被T细胞受体(T cell receptor,TCR)特异性识别。此外,为了验证流程的适用性,我们还对鼠类肉瘤病毒癌基因KRAS与大肠杆菌基因OmpC所编码蛋白质的抗原表位进行了示例性分析。本研究不仅为新冠疫苗设计提供了候选表位,而且还建立了一个广泛适用于多肽疫苗候选表位预测和筛选的操作流程。
1 材料和方法
1.1 数据下载
本研究涉及的SARS-CoV-2的结构蛋白包括刺突糖蛋白(spike glycoprotein,S)、膜糖蛋白(membrane glycoprotein,M)、包膜蛋白(envelope protein,E)与核衣壳蛋白(nucleocapsid phosphoprotein,N),它们的蛋白质序列与鼠类肉瘤病毒癌基因KRAS及大肠杆菌基因OmpC相关的蛋白质序列文件均从NCBI(https://www.ncbi.nlm.nih.gov/)数据库获取,检索ID分别为QHR63290.2、QHD43419.1、QHD-43418.1、QHD43423.2、CAC5395073.1 和 ADU34-074.2。
1.2 表位预测识别
本研究使用生物信息学工具NetMHCpan EL 4.0[8](http://tools.immuneepitope.org/mhci/),对SARSCoV-2的S蛋白、M蛋白、E蛋白与N蛋白及KRAS和OmpC蛋白序列中潜在的T细胞表位分别进行预测。然后,依据“抗原肽-MHC”复合物间的亲和力(通常,%rank<0.5被定义为强结合,%rank<2为弱结合,其他为不结合)进行筛选,保留%rank在0~2的短肽作为结合表位,进一步评估其作为多肽疫苗候选表位的潜力。
1.3 表位免疫原性与抗原性预测
为了筛选出具有免疫原性的候选表位,我们使用IEDB中提供的免疫原性预测工具[6,13](http://tools.iedb.org/immunogenicity/)及本课题组自行开发的INeo-Epp[9](http://www.biostatistics.online/INeo-Epp/)两个生物信息学工具,对S蛋白、M蛋白、E蛋白与N蛋白及KRAS和OmpC蛋白结合表位的免疫原性分别进行预测。IEDB与INeo-Epp工具是通过不同的算法对“抗原肽-MHC”复合物锚定位置处氨基酸的理化特性进行建模,预测效果表现良好[前者的曲线下面积(area under the curve,AUC)=0.69,后者AUC=0.81]。对于免疫原性评分(score),一般认为评分越高表示该表位引发免疫反应的可能性越大[14]。为了降低假阳性率,通常以0.2为标准进行筛选。因此,本研究仅保留IEDB中score大于0.2和INeo-Epp中输出结果为强阳性(positive-high,PH)的表位。
随后,我们使用Vaxijen v2.0[15](http://www.ddgpharmfac.net/vaxijen/)工具预测结合表位的抗原性。Vaxijen是第一个利用非序列比对策略预测表位抗原性的生物信息学工具,也是反向疫苗学中常用的工具。该模型设置了0~1的阈值,以预测病毒、细菌和肿瘤蛋白质序列中的保护性抗原。当阈值为0.4时,该模型在病毒测试数据集的效果较好(AUC=0.74)。因此,本研究在0.4的阈值下获取S蛋白、M蛋白、E蛋白、N蛋白、KRAS蛋白及OmpC蛋白结合表位的抗原性分值。
1.4 表位理化特性分析
为了降低多肽疫苗输注后患者发生不良反应的概率,本研究使用AllerTOP v2.0[16](https://www.ddg-pharmfac.net/AllerTOP/)、ClanTox[17](http://www.clantox.cs.huji.ac.il/)工具对结合表位的理化特性进行分析。AllerTOP和ClanTox拥有非常简洁的操作界面,不需要额外设置阈值即可得到预测结果。此外,AllerTOP还是当前较被认可的预测蛋白质致敏性的工具,它通过自协方差和交叉协方差变换的方法对氨基酸进行编码,综合利用逻辑回归、决策树、随机森林、朴素贝叶斯、多层感知器、K-近邻等算法进行建模,最后选择结果最好的K-近邻算法(AUC=0.85)完成工具的构建。而ClanTox是一种蛋白质毒素分类器,可以根据蛋白质的一级结构计算出该蛋白质是否为毒性蛋白质。在本研究中,我们仅保留AllerTOP和ClanTox输出结果为非致敏(non-allergen)与非毒性(nontoxin)的表位作为SARS-CoV-2多肽疫苗研发的候选表位。
1.5 人群覆盖率评估
根据先前的研究[18~19],我们构建了12种在中国人群中常见的人类白细胞抗原(human leukocyte antigen,HLA)的等位基因(allele)分型。它们分别为HLA-A*01:01、HLA-A*02:01、HLA-A*03:01、HLA-A*11:01、HLA-A*23:01、HLA-A*24:02、HLA-B*07:02、HLA-B*08:01、HLA-B*35:01、HLA-B*40:01、HLA-B*44:02、HLA-B*44:03(表1)。此外,我们使用IEDB提供的人口覆盖率计算工具Population Coverage[20](http://tools.immuneepitope.org/population/)评估了每条候选表位在中国及世界人群中的覆盖率。需要注意的是,使用Population Coverage工具评估表位在特定人群中的覆盖率时,需要提交一份包含该表位所对应HLA等位基因分布频率的信息文件。
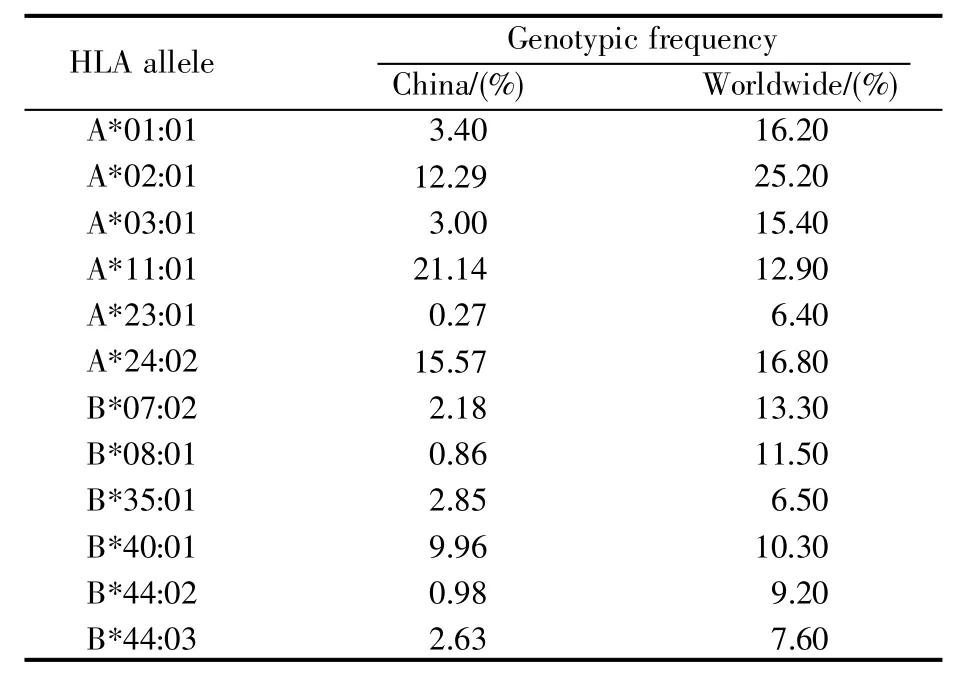
表1 12种HLA等位基因的分布频率Table 1 Distribution frequency of 12 HLA alleles
1.6 同源比对及特异性TCR检索
IEDB[21](http://www.iedb.org/)、VDJdb[22](https://vdjdb.cdr3.net/)、McAPS-TCR[23](http://friedmanlab.weizmann.ac.il/McPAS-TCR/)是当前表位及相关TCR数据储存最多的数据库。首先,本研究收集和整理了3个数据库中有关human、CD8+T、TCRβ的表位信息及其相关的TCR序列信息。然后,应用BLAST(Basic Local Alignment Search Tool)[24](https://blast.ncbi.nlm.nih.gov/Blast.cgi/)工具,将本研究筛选的候选表位与数据库中的表位进行同源比对,记录比对结果中所有同源表位的序列信息及E value和identity值。最后,在已整理的TCR数据中手动检索与该同源表位匹配的TCR序列信息。
2 结果
2.1 SARS-CoV-2结构蛋白中T细胞识别的候选表位
本研究综合多个生物信息学工具(图1A),系统地筛选了SARS-CoV-2结构蛋白序列中可激发T细胞免疫应答的表位,即T细胞表位。首先,我们构建了12种在中国人群普遍存在的HLA等位基因分型:HLA-A*01:01、HLA-A*02:01、HLAA*03:01、HLA-A*11:01、HLA-A*23:01、HLA-A*24:02、HLA-B*07:02、HLA-B*08:01、HLAB*35:01、HLA-B*40:01、HLA-B*44:02、HLAB*44:03;然后,预测了SARS-CoV-2结构蛋白中长度为8~12个氨基酸的T细胞表位。通过“抗原肽-MHC”结合预测工具NetMHCpan EL,共得到117 180条预测表位。之后,根据%rank分值筛选出0~2的结合表位,仅有2 593条表位显示能与MHC分子结合。
免疫原性是指抗原诱导机体发生特异性免疫应答的性能,抗原性则是指抗原与其所诱导产生的抗体或致敏淋巴细胞特异性结合的能力。简而言之,表位的免疫原性与抗原性越强,表明其被T细胞捕获并引发免疫应答的潜力越高。考虑到表位的免疫原性和抗原性均是决定该抗原能否被T淋巴细胞识别的关键特征,我们进一步对结合表位的免疫原性及抗原性进行了分析。首先,使用IEDB和INeo-Epp两个工具分别对2 593条SARS-CoV-2结合表位的免疫原性进行预测,保留在两个工具中结果都较好的表位(IEDB,score>0.2;INeo-Epp,score>0.5即为PH),共获得131条;随后,使用Vaxijen工具分析这131条表位的抗原性,最终仅获得71条既有免疫原性又有抗原性的表位。
疫苗设计的前提是对该病毒候选表位有充分的理解。为此,我们使用蛋白质理化特性分析工具AllerTOP和ClanTox评估了这71条表位的致敏性与毒性,发现其中非致敏和非毒性的候选表位有49条。此外,由于HLA等位基因的多态性,我们发现候选表位中存在同一表位与多个HLA等位基因结合的情况,例如:表位M135~144和表位S1110~1121以不同的%rank 分值与 HLA-B*40:01、HLA-B*44:02及HLA-B44:03三种分型结合。候选表位与HLA等位基因分型的结合率如图1B所示。
最后,在忽略同一表位与不同HLA等位基因结合的情况下,我们仍在49条表位中找到了34条独特的表位(表2),其中15条来自S蛋白序列、9条来自M蛋白序列、5条来自E蛋白序列、5条来自N蛋白序列(图1C)。总体来看,这34条候选表位经过了系统和严格的筛选,可以与MHC分子结合,并由抗原提呈细胞加工后呈递到细胞表面,供TCR识别,继而诱导T细胞发挥效应功能。

表2 SARS-CoV-2的候选表位信息Table 2 Information about candidate epitopes of SARS-CoV-2

图1 多肽疫苗候选表位预测流程及其在SARS-CoV-2中的应用(A)SARS-CoV-2 T细胞候选表位的预测流程图;(B)候选表位与HLA等位基因的结合率;(C)不同预测过程中得到的SARSCoV-2表位数量。Fig.1 The workflow for predicting candidate epitopes for peptide vaccines and its application in SARS-CoV-2(A)Screening process of SARS-CoV-2 T cell candidate epitopes;(B)Binding rates of candidate epitopes and HLA alleles;(C)The number of SARS-CoV-2 epitopes retained during the screening process.
2.2 候选表位的人群覆盖率
由于与表位结合的HLA等位基因在不同国家的分布频率差异明显,我们评估了SARS-CoV-2的34条候选表位在中国及世界范围的覆盖率。首先,我们获取了HLA等位基因在中国及世界范围内的分布频率[18~19]。然后,基于HLA等位基因的分布频率,使用Population Coverage工具计算了每条候选表位在中国和世界人群的覆盖率。结果显示,本研究筛选的SARS-CoV-2候选表位在中国人群广泛覆盖(1%~91%)(表2)。其中,表位S1110~1121与表位 M135~144在中国人群的适用范围高达91%,同时在世界人群的适用范围也达到71%。
2.3 同源表位及其诱导的TCR序列
为了检验本研究所构建预测流程的实用性,我们使用BLAST工具将本文筛选的34条SARSCoV-2表位与数据库中经过验证的表位进行同源比对。结果显示,有20条SARS-CoV-2候选表位与 IEDB、VDJdb、McAPS-TCR 3个数据库中经过验证的表位同源(表3)。另外,当比对结果的参数E value调整为 0.05 时,E29~38和 E30~37这两条表位与数据库中LLAILTYYV(blast epitope)表位的identity高达100%。与此同时,我们还在数据库中检索到6条与该同源表位相关的特异性TCR序列(CASSLVRDRHTEAFF、CASSPTGTGGSDTQYF、CASSQAGEQYF、CASSWVGGADTQYF、CASTVRQGSNQPQHF 和 CSASFHNGFWGGTEAFF)。
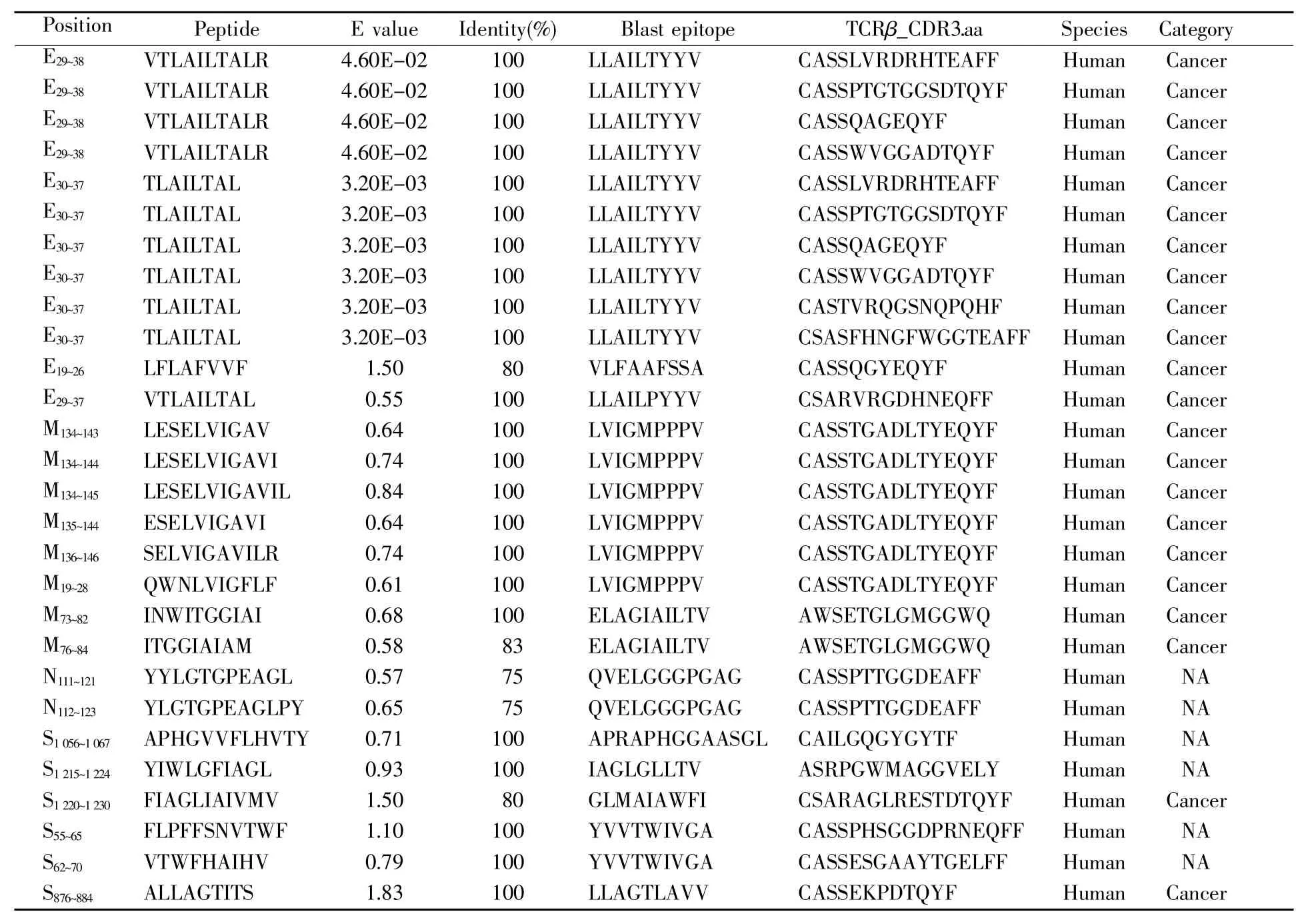
表3 SARS-CoV-2的同源表位及相关TCR序列信息Table 3 Information about homologous epitopes and relevant TCR sequences of SARS-CoV-2
2.4 其他病原体蛋白质序列中候选表位的预测与筛选
为了进一步验证该流程的适用性,我们在NCBI中检索了鼠类肉瘤病毒癌基因KRAS及大肠杆菌基因OmpC相关的蛋白质序列,并对蛋白质序列中适合多肽疫苗研发的表位进行了预测和筛选。从表4可知,本研究共预测到5条有关KRAS的表位,即 KLVVVGAGGVGK、VVVGAGGVGK、LVVVGAGGVGK、QYMRTGEGFL和YMRTGEGF。基于这5条表位的%rank分值、免疫原性、抗原性、致敏性及毒性,我们发现7~16区段的表位VVVGAGGVGK有潜力成为KRAS多肽疫苗研发的候选表位。另外,从表5的同源比对结果可以看出,VVVGAGGVGK与数据库中YMDDVVLGA表位的identity达到80%,同时还有一条特异性TCR序列CASSYLTGEGDYGYTF与该同源表位相关,这进一步表明表位VVVGAGGVGK具有免疫原性,能够诱导T细胞免疫应答。

表4 KRAS的候选表位信息Table 4 Information about candidate epitopes of KRAS

表5 KRAS的同源表位及相关TCR序列信息Table 5 Information about homologous epitopes and relevant TCR sequences of KRAS
我们在基因OmpC相关的蛋白质序列中共预测到12条表位(表6)。从预测表位的各预测分值及同源比对结果可知,表位YEGFGIGGAI(205~214)、VLPEFGGDTY(127~136)、TDVLPEFGGDTY(125~136)、DVLPEFGGDTY(126~136)可以作为OmpC多肽疫苗研发的候选表位(表6~7)。而且,对比分析 125~136、126~136、127~136 区段可以发现,125~136区段是OmpC蛋白中适用于多肽疫苗研发的优势区段。由此,我们认为本研究所提出的预测流程可适用于病原体抗原蛋白序列中多肽疫苗候选表位的预测和筛选。

表6 OmpC的候选表位信息Table 6 Information about candidate epitopes of OmpC

表7 OmpC的同源表位及相关TCR序列信息Table 7 Information about homologous epitopes and relevant TCR sequences of OmpC
3 讨论
快速、安全、灵活、高效且低成本的新型疫苗研发路线是未来人类对抗病原体最有效的手段[25]。近年来,随着生物信息学技术的发展和应用,研究人员已经可以从病原体高通量组学信息中挖掘出大量的免疫原性表位序列,同时实现对T/B细胞靶点处表位的精准识别,从而为新型疫苗的研发提供理论指导[26]。基于此,借助生物信息学工具初筛病原体蛋白质序列中的候选表位已经成为疫苗研究中关键的一步。在此次新型冠状病毒肺炎(COVID-19)发生之初,Grifoni团队[19]就使用Net-MHCpan EL web工具对SARS-CoV-2蛋白质序列中潜在的T、B细胞表位进行了预测,经同源比对发现,这些表位与IEDB中记录的SARS表位有较高的同源性(B细胞表位为69%~100%;T细胞表位为17%~100%)。由此,我们综合利用当前的生物信息学工具,对SARS-CoV-2结构蛋白中潜在的T细胞候选表位进行了预测和筛选,共获得34条符合MHCⅠ类分子的T细胞候选表位信息,其中候选表位 S1110~1121和 M135~144覆盖高达91%的中国人群和71%的世界人群,可为COVID-19的疫苗设计提供参考。
目前,利用机器学习算法,相关人员已经开发了系列针对抗原提呈过程的生物信息学工具。比如,利用人工神经网络算法开发的NetMHCpan系列工具,既拥有操作简洁的web页面,也有适用于Linux系统的版本。另外,本课题组利用随机森林算法开发的INeo-Epp工具可以预测抗原肽的免疫原性,其AUC达到0.81,而IEDB中提供的同类型工具的AUC仅为0.69。除本研究中涉及的工具外,还有诸多类似功能的工具可用于疫苗候选表位的预测和筛选,例如:NetCTL 1.2(http://www.cbs.dtu.dk/services/NetCTL/)可用于预测蛋白质序列中潜在的细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTL)表位[27];MixMHC2pred[28]可用于预测HLAⅡ类分子呈递的抗原肽。这些生物信息学预测工具在抗原特定功能的预测中均显示出良好的性能,但在实现疫苗候选表位的完整预测和分析方面仍存在一定难度。
综上所述,本研究综合利用多种生物信息学工具,结合自主研发的算法工具,整理并提出了一个可广泛应用于病原体多肽疫苗候选表位预测和分析的流程。通过该工作流程可以预测出不同抗原蛋白质序列中高可信度的疫苗候选表位,从而为预防和控制感染性疾病与恶性肿瘤的新型疫苗的研制提供参考。
