镍催化的吲哚2-位二氟烷基化反应的研究
2021-09-01赖胤龙张洁蓥李健华严绍熙杜克斯罗建民
赖胤龙, 张 琦, 张洁蓥, 李健华, 严绍熙, 杜克斯, 罗建民*
(1. 韶关学院 化学与土木工程学院,广东 韶关 512005; 2. 华南理工大学 环境与能源学院,广东 广州 510655)
吲哚结构是一类许多天然化合物和合成生物活性分子中重要的核心骨架。将不同基团引入吲哚骨架进行结构修饰可以合成一系列具有生物活性的吲哚衍生物[1]。此外,鉴于二氟官能团(CF2)具有特殊化学和生理学性质,在特定位置引入二氟官能团,不仅可以保持原有的化合物的生物活性,还能改善其溶解性、脂溶性、代谢稳定性、pKa及构象等性质[2-3]。因此,将二氟官能团选择性地引入到吲哚化合物中是一种重要的合成方法,已被公认为药物开发的有力策略。
近年来,利用过渡金属催化C—H键的氟烷基化反应已获得了较好的发展,尤其是钌、铑、钯、铜、镍等过渡金属催化吲哚的区域选择性二氟烷基化反应[4-12]。如Qing课题组和Wang课题组分别在光化学条件下,以钌金属盐为催化剂,得到了适中收率的2-二氟甲基吲哚衍生物(Scheme 1a)[4-5]。接着,Zhang课题组以金属钯为催化剂实现了直接的C—H-二氟甲基化反应,反应中双齿膦配体配体(Xantphos)起到了的关键作用(Scheme 1b)[6]。随后,Huang课题组和Wang课题组在高温加热条件下通过当量或催化量的铜盐完成了吲哚二氟化和全氟烷基化反应(Scheme 1c)[7-8]。 2017年,Loh课题组报道了一种铑催化具有N-中心2-嘧啶基作为引导基团的吲哚2-位的C—H选择性二氟烷基化反应(Scheme 1d)[9]。最近,Punji课题组在高温加热的(DME)NiCl2/Xantphos催化体系中,实现了N-取代吲哚的区域选择性的二氟烷基化反应(Scheme 1e)[10]。综上所述,构建2-二氟烷基吲哚的报道相对较少。
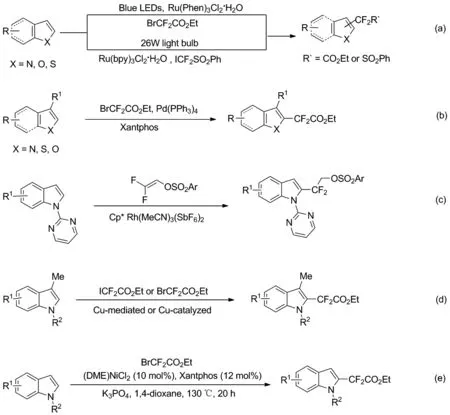
Scheme 1
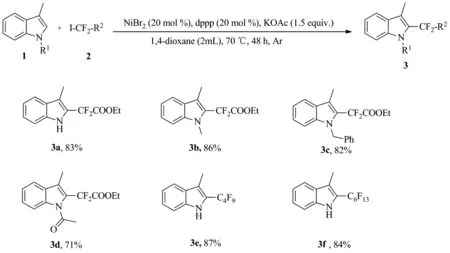
Scheme 2

Scheme 3
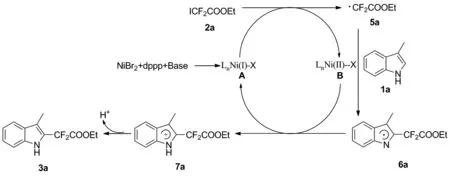
Scheme 4
因此,寻找一种更加温和条件下的廉价镍催化直接吲哚2-位C—H键的二氟烷基化反应在有机氟化学合成上具有重要的意义。在本课题组前期研究成果基础上[11-12],本文尝试采用便宜的NiBr2为催化剂,dppp为配体,实现了吲哚2-位C—H键的二氟烷基化反应。
1 实验部分
1.1 仪器与试剂
Bruker 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标)。
所用试剂均为分析纯。
1.2 3a~3f的合成(以3a为例)
在干燥的Schlenk管中加入3-甲基吲哚1a(0.3 mmol)、 NiBr2(0.06 mmol)、 dppp(0.06 mmol)和KOAc(0.45 mmol),然后抽换氮气 3 次,用注射器分别加入碘二氟乙酸乙酯2a(0.3 mmol)和溶剂1,4-dioxane(2 mL),于70 ℃反应至终点(TLC跟踪)。用乙酸乙酯(3×10 mL)萃取,合并有机相,依次用15 mL饱和食盐水洗涤,无水硫酸镁干燥,过滤,滤液旋蒸除去有机溶剂,残余物经硅胶柱层析纯化得化合物3a。 用类似的方法合成3b~3f。
2-(3-甲基-1H-吲哚-2-基)-2,2-二氟乙酸乙酯(3a)[4]: 黄色液体,收率83%;1H NMR(400 MHz, CDCl3)δ: 8.47(br s, 1H), 7.66(d,J=8.0 Hz, 1H), 7.39(d,J=8.0 Hz, 1H), 7.32(t,J=8.0 Hz, 1H), 7.22~7.19(m, 1H), 4.37(q,J=8.0 Hz, 2H), 2.48~3.47(m, 3H), 1.36(t,J=8.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 163.68(t,J=36.0 Hz), 135.78, 128.71, 124.39, 123.55(t,J=30.0 Hz), 120.22, 119.93 113.99(t,J=4.0 Hz), 111.66, 111.63(t,J=250.0 Hz), 63.65, 14.04, 8.60;19F NMR(376 MHz, CDCl3)δ: -102.01(s, 2F)。
2-(1,3-二甲基-1H-吲哚-2-基)-2,2-二氟乙酸乙酯(3b)[6]: 黄色液体,收率86%;1H NMR(400 MHz, CDCl3)δ: 7.68(d,J=8.0 Hz, 1H), 7.38~7.37(m, 2H), 7.23~7.19(m, 1H), 4.37(q,J=8.0 Hz, 2H), 3.89(s, 3H), 2.51(t,J=4.0 Hz, 3H), 1.38(t,J=8.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 163.81(t,J=36.0 Hz), 138.20, 127.75, 124.96(t,J=29.0 Hz), 124.16, 119.95, 119.78, 114.40(t,J=4.0 Hz), 112.57(t,J=250.0 Hz), 109.66, 63.51, 31.51(t,J=4.3 Hz), 14.00, 9.07(t,J=3.0 Hz);19F NMR(376 MHz, CDCl3)δ: -98.60(s, 2F)。
2-(1-苄基-3-甲基-1H-吲哚-2-基)-2,2-二氟乙酸乙酯(3c)[6]: 黄色固体,收率82%;1H NMR(400 MHz, CDCl3)δ: 7.77(d,J=8.0 Hz, 1H), 7.35~7.25(m, 6H), 7.03(d,J=8.0 Hz, 2H), 5.63(s, 2H), 4.19(q,J=7.2 Hz, 2H), 2.62~2.61(m, 3H), 1.30(t,J=8.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 163.57(t,J=36.0 Hz), 138.04, 128.65, 128.06, 127.24, 125.84, 125.05(t,J=29.0 Hz), 124.48, 120.18, 120.04, 115.08(t,J=4.0 Hz), 112.54(t,J=250.0 Hz), 110.54, 63.47, 48.33(t,J=4.0 Hz), 13.86, 9.27(t,J=3.0 Hz);19F NMR(376 MHz, CDCl3)δ: -98.32(s, 2F)。
2-(1-乙酰基-3-甲基-1H-吲哚-2-基)-2,2-二氟乙酸乙酯(3d)[7]: 黄色固体,收率71%;1H NMR(400 MHz, CDCl3)δ: 7.64(t,J=8.0 Hz, 2H), 7.43(t,J=8.0 Hz, 1H), 7.34(t,J=8.0 Hz, 1H), 4.37(q,J=8.0 Hz, 2H), 2.77(s, 3H), 2.49(t,J=4.0 Hz, 3H), 1.36(t,J=8.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 168.99, 163.00(t,J=33.0 Hz), 135.13, 131.22, 126.71, 126.66(t,J=29.0 Hz), 124.19(t,J=3.0 Hz), 123.49, 120.77,113.97, 112.97(t,J=250.0 Hz), 62.77, 26.70, 14.07, 9.84(t,J=6.0 Hz);19F NMR(376 MHz, CDCl3)δ: -96.4 0(s, 2F)。
3-甲基-2-(全氟丁基)-1H-吲哚(3e)[7]: 白色固体,收率87%;1H NMR(400 MHz, CDCl3)δ: 8.05(br s, 1H), 7.61(d,J=8.0 Hz, 1H), 7.31~7.28(m, 2H), 7.18~7.14(m, 1H), 2.39(t,J=4.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 136.19, 128.58, 125.14, 120.58, 120.29, 119.66(t,J=29.0 Hz), 116.95(t,J=4.0 Hz), 111.72, 8.66(t,J=1.0 Hz);19F NMR(376 MHz, CDCl3)δ: -81.47~-81.53(m, 3F), -109.27~-109.34(m, 2F), -123.46~-123.54(m, 2F), -126.41~-126.53(m, 2F)。
3-甲基-2-(全氟己基)-1H吲哚(3f)[7]: 白色固体,收率84%;1H NMR(400 MHz, CDCl3)δ: 8.16(br s, 1H), 7.67(d,J=8.0 Hz, 1H), 7.41~7.33(m, 2H), 7.23~7.20(m, 1H), 2.46(s, 3H);13C NMR(100 MHz, CDCl3)δ: 136.29, 128.67, 125.16, 120.61, 120.32, 119.86(t,J=28.0 Hz), 117.02(t,J=3.0 Hz), 111.75, 8.57;19F NMR(376 MHz, CDCl3)δ: -81.27~-81.33(m, 3F), -109.04~-109.12(m, 2F), -122.30~-122.37(m, 2F), -122.56~-122.67(m, 2F), -123.16~-123.26(m, 2F), -126.53~-126.64(m, 2F)。
2 结果与讨论
2.1 反应条件优化
以3-甲基吲哚(1a)和碘二氟乙酸乙酯(2a)为模版底物,进行反应条件优化的研究(表1)。由表1可知,当在反应温度70℃,Ni(OAc)2(0.06 mmol)作催化剂,dppp(0.06 mmol)作配体,KOAc(0.45 mmol)作碱,在1,4-dioxane(2 mL)溶剂中,氩气条件下反应48 h,发现可获得18%的目标产物3a(Entry 1)。接下来研究了不同的镍化合物的催化效率。结果表明,当用NiBr2作为催化剂时,得到最高的收率(Entries 2~5)。随后进行了配体的筛选,发现dppe和dppf的作用不如dppp有效(Entries 6~7)。接着探讨不同的碱(如K3PO4、 K2CO3和KHCO3)对反应的影响,结果显示KOAc的效果最优(Entries 8~10)。在此基础上筛选了不同的反应溶剂,如二甲基亚砜(DMSO)、N,N-二甲基甲酰胺(DMA)、乙腈(CH3CN)和四氢呋喃(THF),结果表明采用1,4-dioxane作溶剂,收率最高(Entries 11~14)。在反应温度方面,70 ℃的收率最高(Entries 15~16)。
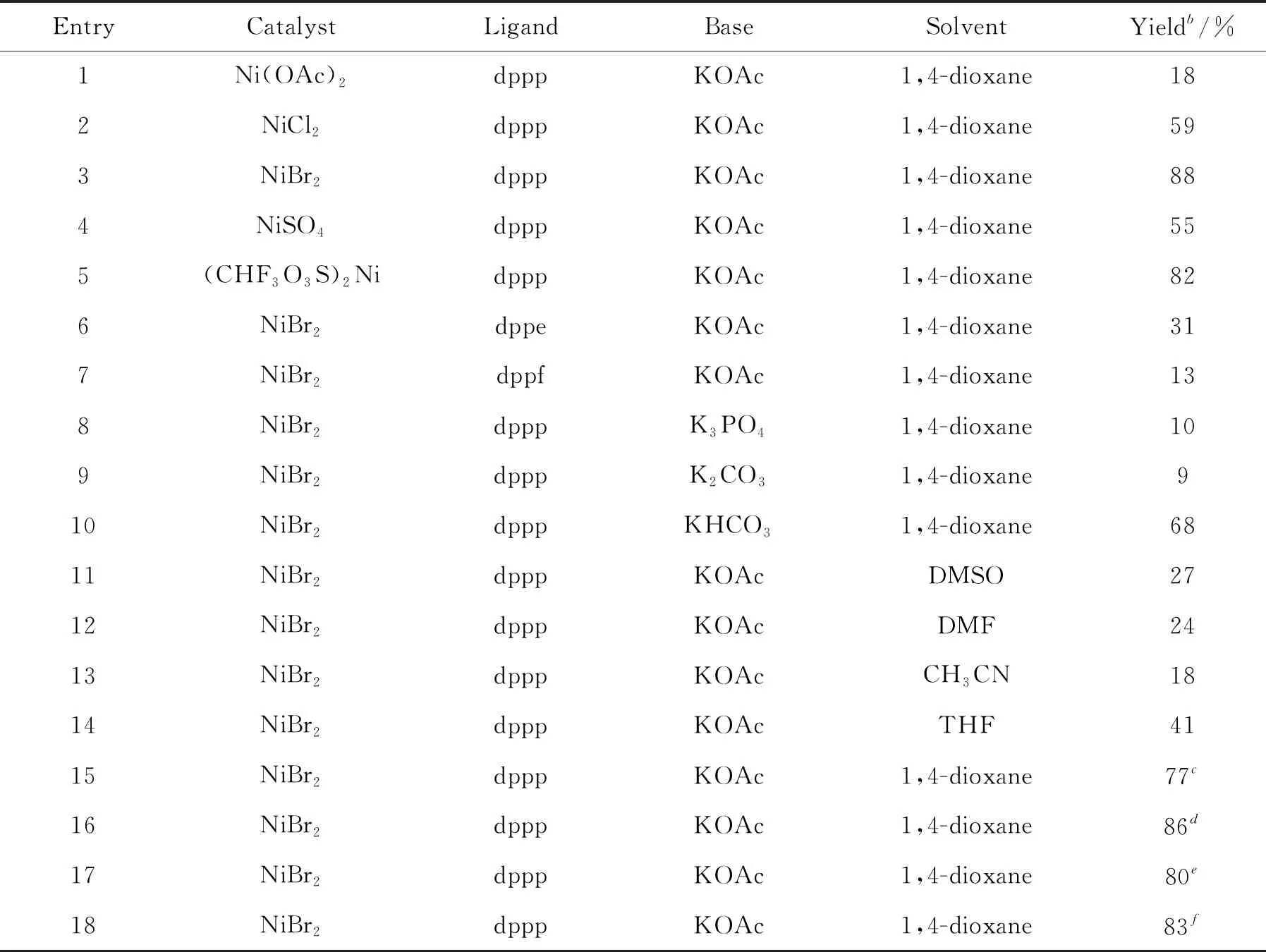
表1 反应条件的优化
最后研究了不同时间对反应收率的影响(24 h和72 h),最终发现48 h的收率最理想(Entries 17~18)。
2.2 底物拓展
在最佳反应条件下,进行了底物适用性的研究,如图Scheme 2所示。实验结果发现当吲哚底物具有甲基和苄基等供电子基团时,反应可以得到较好的收率,如产物3b和3c。而当吲哚底物具有羰基这样的吸电子基团时,收率略有下降,如产物3d。烷基氟基团在药物合成中是一类重要的生物电子等排基团,因此,也对烷基氟试剂进行了考察。实验表明3-甲基吲哚可以和全氟碘代烷反应,而且收率较高,如产物3e和3f。
2.3 控制性实验
为了探究反应的机理,进行了控制性实验,如Scheme 3所示。首先在标准条件下,把自由基抑制剂TEMPO(2.0 eq.)或BHT(2.0 eq.)加入到反应体系中,发现该反应被完全抑制,只获得了trace的收率(eq 1)。这暗示了反应可能是一个自由基的过程;随后,采用自由基捕获剂(1,1-二苯乙烯)捕获·CF2COEt自由基,发现·CF2COEt自由基被成功捕获,4a收率67%(eq 2)。
2.4 可能的反应机理
根据控制性实验和文献[10,14-15]调研结果,分析了一种可能的反应的机理,结果见Scheme 4:首先,溴化镍、dppp和碱共同作用现场形成一价镍中间体A,该中间体A随后与碘二氟乙酸乙酯2a反应生成·CF2COEt自由基5a和二价镍中间体B,接着·CF2COEt自由基5a与3-甲基吲哚反应生成吲哚自由基中间体6a,然后二价镍中间体氧化生成吲哚正离子中间体7a,最后吲哚正离子中间体7a脱去一个氢生成目标产物3a。
报道了一种用廉价镍催化的N-取代或不取代吲哚2-位的二氟烷基化和全氟烷基化反应。反应在温和的条件下具有高收率和良好底物适用性。机理研究表明,反应可能包含了一个自由基的途径。
