人工模拟条件下环境DNA 宏条形码技术的定量分析初探*
2021-08-25赵新宁柳淑芳庄志猛
牟 铭 李 昂 赵新宁 柳淑芳① 庄志猛
(1. 上海海洋大学水产与生命学院 上海 201306;2. 中国水产科学研究院黄海水产研究所 青岛海洋科学与技术试点国家实验室海洋渔业科学与食物产出过程功能实验室 山东 青岛 266071)
环境 DNA 宏条形码技术(environmental DNA metabarcoding)可以通过高通量测序对环境 DNA(eDNA)进行多物种分类鉴定。随着eDNA 宏条形码技术的应用和发展,该技术对群落中单一或组合物种的定量评估逐渐显现出优势。无论是水族箱或中观实验等小型生态系统(Minamotoet al; Moyeret al, 2014;Pilliodet al, 2013; Thomsenet al, 2012; Doiet al,2017),还是江河湖泊等自然淡水系统(Doiet al, 2017;Lacoursiererousselet al, 2016),甚至是开放式的海洋系统(Joet al, 2017),研究人员都尝试过使用eDNA宏条形码技术测算种群相对数量。大量研究表明,特定环境中物种的个体数量和基于eDNA 宏条形码技术获得的高通量测序reads 数之间具有明显的相关性。Evans 等(2016)设计了一个包含鱼类和两栖类的中观实验,证明水体中物种个体数或生物量相对丰度与eDNA 宏条形码技术获得的高通量测序reads 数呈正相关;Hanfling 等(2016)对比较了一个天然湖泊的长期监测鱼类丰度结果与eDNA 宏条形码技术得到的高通量测序reads 数之间的相关性,结果表明二者呈正相关;Thomsen 等(2016)研究发现,当将鱼类的eDNA 高通量测序reads 数聚类到“科”的分类等级时,eDNA 宏条形码技术测得的深海生境中的生物量与传统拖网捕捞数据比较吻合。可以预见,eDNA 宏条形码技术具有快速测算群落中物种丰度的潜能,将成为资源保护和管理中具有应用前景的调查工具。
然而,通过对已发表的相关文献进行梳理和归纳,发现当前大多数利用eDNA 高通量测序结果的reads 数来评估自然环境中生物相对数量的研究并不能得到明确的量化关系结果(Limet al, 2016)。究其原因,是因为从生物到eDNA 以及从eDNA 再到高通量测序结果的2 个过程中的数学关系难以理清。其中,实验室操作过程中的主要问题是:(1)自然水体中,不同浓度 eDNA 富集效率不同(Kellyet al, 2016);(2)PCR 扩增过程中,通用引物对不同物种DNA 存在偏倚性(Elbrechtet al, 2015; Pinolet al, 2015)。假定水体中的eDNA 全部回收,且PCR 扩增时不存在引物偏倚性,这种理想状态下的水体中eDNA 组成与其高通量测序reads 数是否完全呈线性关系?为了探究这个问题,本研究在实验室可控条件下,选择2 个同属近缘种,对其DNA 样品进行不同比例混合,模拟从自然水体中富集到的eDNA 复合样品,既保证了样品的回收率,又降低了引物偏倚的干扰。以此为模板,探究eDNA 宏条形码技术检测种群相对数量的准确性,旨在为验证eDNA 宏条形码技术监测水生生物资源量的可行性提供直接证据。
1 材料与方法
1.1 实验方案设计
1.1.1 实验样品的选择 鉴于通用引物对不同物种的扩增效率有所不同,本研究实验样品选取2 个同属近缘物种用于制备模拟eDNA 样品,以确保将PCR过程中因引物对物种DNA 的偏移引起的实验误差降低到最小程度。
实验样品选择了2 种常见对虾:凡纳滨对虾(Penaeus vannamei)和墨吉对虾(Penaeus merguiensis),它们同为节肢动物门(Arthropoda)、软甲纲(Malacostraca)、十足目(Decapoda)、对虾科(Penaeidae)、对虾属(Penaeus),是较为常见的同属近缘种。
1.1.2 模板DNA 的提取 采用传统的酚-氯仿-异戊醇方法分别提取2 种对虾肌肉组织DNA,经纯化后使用超微量核酸蛋白分析仪(GE,美国)检测浓度,稀释DNA 浓度至20 ng/μL (±1%),-20℃保存备用。
1.1.3 模拟eDNA 的制备 人工模拟eDNA 复合样品,共设7 个实验组:将凡纳滨对虾和墨吉对虾的DNA 模板分别按照1∶1、1∶2、1∶4、1∶8、1∶10、1∶50 和1∶100 的比例均匀混合,且混合后DNA 终浓度一致。设阳性对照2 组:分别选用凡纳滨对虾和墨吉对虾DNA 样品。设阴性对照1 组:用纯水代替DNA 样品。每组样品设3 个重复。
将上述制备的eDNA 复合样品作为eDNA 宏条形码检测的模板DNA。
1.2 通用引物的选取
考虑到后续高通量测序及DNA 条形码数据库信息资源的丰富度,本研究选取了Leray 等(2013)推荐的长度为313 bp 的线粒体COⅠ基因序列作为待测eDNA 目标片段,该片段长度适中、数据资源丰富、物种鉴定特异性好,且引物通用性强。通用引物序列见表1,由华大基因有限公司合成。
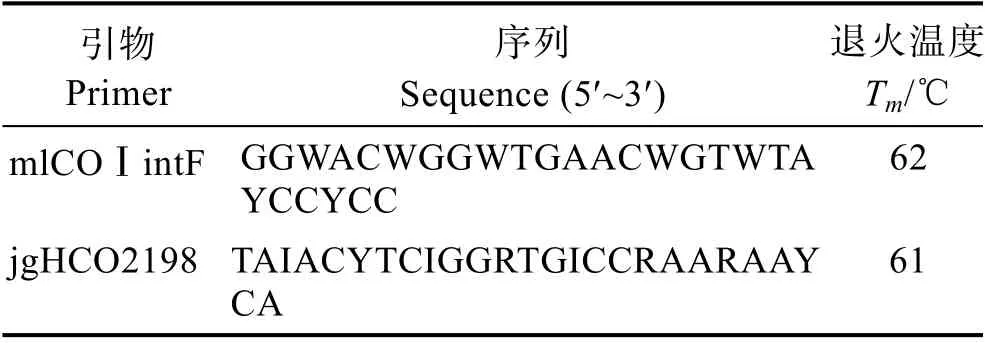
表1 COⅠ基因PCR 扩增引物信息Tab.1 Primer sets of COⅠ used for PCR amplification
1.3 PCR 扩增
PCR 扩 增 体 系:2×RapidTaqMaster Mix(Vazyme) 25 μL,上下游引物(mlCOⅠintF/jgHCO2198)(10mmol/L)各2 μL,DNA 模板5 μL,无菌水补齐总体积至50 μL。
为了增强扩增特异性,使用Touch down PCR 反应程序:95℃预变性10 min;95℃变性30 s,62℃退火30 s,72℃延伸30 s,16 个循环;95℃变性30 s,50℃退火30 s,72℃延伸30 s,25 个循环;72℃延伸3 min;4℃保存。
用1%琼脂糖凝胶电泳检测PCR 产物质量,选用DL2000 DNA marker,目标条带约为300 bp。挑选电泳条带单一且明亮的PCR 产物用于高通量测序。
1.4 测序及结果注释
将所得PCR 产物送至青岛欧易生物科技有限公司,进行高通量测序。采用Illumina Miseq 平台测序。
按照97%相似性对非重复序列(不含单序列)进行OTU(operational taxonomic units)聚类,在聚类过程中去除嵌合体(chimera,融合了2 个不同序列信息的序列)、异源双链序列等干扰序列,得到OTU 聚类结果,然后对不同OTU 的代表序列进行结果注释。在结果注释时,参照 NCBI 公共数据库(https://www.ncbi.nlm.nih.gov/)来进行分类学信息的标注,同时使用中国重要渔业生物DNA 条形码信息平台(http://www.fisherybarcode.cn)对注释结果进行辅助校正。
1.5 数据处理
统计每个样品高通量测序的原始reads 数,以所有样本中reads 数最低值为基准,进行抽平,使每个样品测序结果的reads 数相同,用于后续数据分析。
在推导数学关系时,设:eDNA 样品来自2 个物种,分别为A、B;实际参与PCR 反应的底物模板DNA 的量为n;参与PCR 反应的底物模板DNA 理论量为x;每个循环中,参与扩增的2 个物种底物模板DNA 比率为λ;扩增的循环数为c;PCR 产物的高通量测序reads 数为y。假定理想情况为:在PCR 扩增的不同循环中,参与扩增的2 个物种底物模板DNA比率固定;PCR 扩增全程都保持底数为2 的指数增长型(反应物足够),则高通量测序reads 数可以反应eDNA 样品中2 个物种的相对量,即推得公式(1):
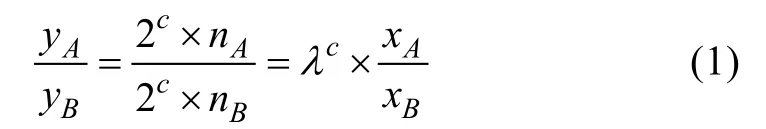
经变形得公式(2):

2 结果与分析
2.1 PCR 扩增结果
以人工模拟的7 组eDNA 复合样品及对照组样品为模板,使用eDNA 宏条形码通用引物进行PCR 扩增,经1%琼脂糖凝胶电泳检测,结果显示,阳性对照和7 个实验组均成功扩增出300 bp 左右的目标片段,阴性对照无扩增产物(图1)。

图1 琼脂糖凝胶电泳检测结果Fig.1 Results of agarose gel electrophoresis
2.2 测序数据
2.2.1 高通量测序结果 本研究对7 个实验组共21 个PCR 产物进行了高通量测序。统计高通量测序获得的序列长度分布,显示99.80%以上序列的长度分布于301~350 bp 之间(图2)。抽平后共计获得有效序列674,205 条,序列平均长度为312.41 bp。

图2 测序数据长度分布统计Fig.2 Result of distributional interval of the sequence length
2.2.2 分类学信息注释及reads 数统计结果 将每个样品reads 总数抽平后,对674,205 条序列进行分类学信息注释,保留有效序列(序列长度在301~350之间,且分类学信息注释结果是墨吉对虾和凡纳滨对虾)的reads。各实验组高通量测序reads 数及其分类注释结果详见表2。实验组I 的3 组平行样共获得94,774 条序列,每个样品注释出的墨吉对虾和凡纳滨对虾平均reads 数分别为11,097 和20,494,二者的比值为13/24;实验组Ⅱ的3 组平行样共获得95,208 条序列,二者reads 数比值为18/23;实验组Ⅲ的3 组平行样共获得 95,572 条序列,二者 reads 数比值为77/73;实验组Ⅳ的3 组平行样共获得95,250 条序列,二者reads 数比值为67/44;实验组Ⅴ的3 组平行样共获得94,408 条序列,二者reads 数比值为43/24;实验组Ⅵ的3 组平行样共获得94,640 条序列,二者reads数比值为 127/35;实验组Ⅶ的 3 组平行样共获得95,526 条序列,二者reads 数比值为187/23。

表2 各实验组获得的序列数及分类学信息注释Tab.2 Results of taxonomic information and numbers of reads
2.2.3 混合模板PCR 扩增的引物偏倚率 当模板中2 个物种DNA 浓度比例为1∶1 (墨吉对虾/凡纳滨对虾)时,2 个物种的模板DNA 含量相同,扩增过程中主要是引物对不同底物的偏好引起的扩增产物差别,此时主要是引物偏倚引起的reads 数差距。根据公式(2)变形得:

代入扩增循环数,由reads 平均数比值,计算得到每个循环中参与反应的2 个物种模板DNA 的平均比率为66/67,换言之,PCR 扩增的每个循环中,平均偏倚率为1/67,约为1.5%,即该引物在同一PCR体系中,每次退火过程对墨吉对虾DNA 模板的结合率比凡纳滨对虾DNA 模板大约少1.5%。
2.2.4 eDNA 组成与高通量测序reads 数的函数关系
以各实验组设置的底物中墨吉对虾DNA 与凡纳滨对虾DNA 的比值为横坐标,以各实验组样品高通量测序结果注释的墨吉对虾与凡纳滨对虾reads 平均数的比值为纵坐标,绘制eDNA 组成与高通量测序reads 数的函数关系图。在公式(2)的前提假设条件下,在高通量结果中,2 种对虾的reads 数之比和初始PCR模板DNA 中2 种对虾的DNA 比例应呈现y=kx线性关系,对表2 中的7 个实验组的reads 平均数比值和PCR 模板DNA 比值的2 组数据进行一元线性回归分析,最终得出eDNA 组成与高通量测序reads 数的函数关系:y=0.0716x+0.7043 (r²=0.9824)(图3)。
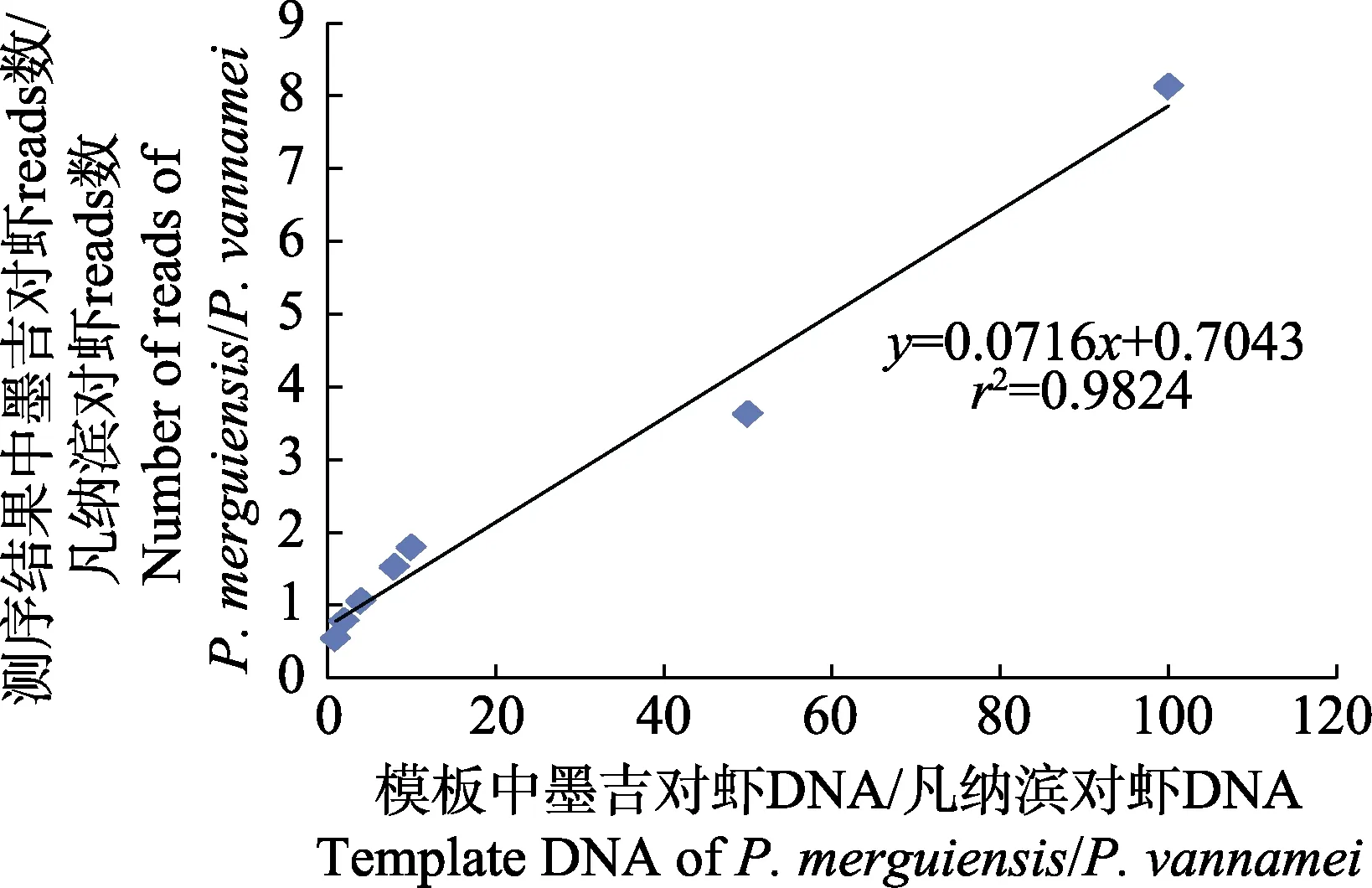
图3 2 个物种高通量测序结果的序列数比值与模板比例的关系Fig.3 The curve of ratio of two species'reads and ratio of template DNA
3 讨论
利用eDNA 宏条形码技术来评估水体中不同物种的相对数量,是该技术最具潜力的应用前景之一。与传统调查方法相比,eDNA 宏条形码技术具有操作简便、省时高效且对自然资源破坏小等特点。如何使用该技术来监测水体中的生物量,一直是学界关注的焦点,但水体中的eDNA 难以完全富集,且富集到的eDNA 成分复杂;PCR 扩增时,引物对不同种类DNA模板的偏好还会导致扩增效率不同,这些问题亦是eDNA 宏条形码技术推广应用的最大障碍。为运用eDNA 宏条形码技术检测环境生物相对数量而设计的中观实验,大都是在尝试推导生物量与高通量测序结果reads 数的数学关系,主要包括环境中生物量与eDNA 量、环境中eDNA 量与提取的eDNA 量和提取的eDNA 模板与PCR 产物的高通量测序结果等3 个对应关系,其中任何一个关系不明确,都将影响eDNA 宏条形码技术的检测结果,所以,必须逐步理清这3 种对应关系的函数关系,进而得到“高鲁棒性”(模式体系固定且成熟,效果准确且稳定)的eDNA宏条形码方法应用体系。
分解困难是解决问题的有效方法之一,剖析DNA宏条形码技术全过程,当只关注富集到的eDNA 扩增及测序时,如果能创造一种eDNA 成分简单、无引物偏好影响的理想条件,进而探索eDNA 高通量测序reads 数和底物eDNA 含量之间的数学关系,便可以收获“柳暗花明”的结果。
3.1 混合模板PCR 扩增的引物偏倚性
影响多物种eDNA 混合模板PCR 扩增效率的主要原因之一,便是PCR 过程中引物对不同模板DNA碱基组成存在偏好性(Kanagawa, 2003)。理论上,引物的每个脱氧核苷酸和模板DNA 链上同源区段的每个脱氧核苷酸可以实现一一对应的关系,但实践过程中通常难以实现,任何一个错配位点的出现都会导致相应的扩增效率降低,从而减少产物。为此,通用引物中加入了简并碱基以降低错配的影响。然而,在进行复杂模板的PCR 扩增时,即使使用含简并碱基的通用引物,引物结合能力也会因为不同模板DNA 引物结合区段序列的GC 碱基含量高低而不同(Acinaset al, 2005),GC 含量高的引物结合力较强(Fonseca,2018)。为了提高模板DNA 引物结合区段碱基组成的一致性,从而降低引物偏倚的影响,本研究选择较为常见的同属近缘种凡纳滨对虾和墨吉对虾作为实验对象。同时,基于基因的进化速率及其数据库信息的丰富程度等因素,决定以mtDNA 中的COⅠ基因片段作为检测的目的基因。
通过控制上述引物偏好的条件,最终高通量测序结果中引物的偏倚程度如何。研究分析发现,当eDNA 模板中凡纳滨对虾和墨吉对虾DNA 组成比例为1∶1 时,高通量测序获得对应物种reads 数比例为13/24。根据扩增循环数和公式(2),推导此时参与扩增的2 种DNA 比约为66/67,即PCR 扩增的平均偏倚率为1/67,约1.5%。可见选择凡纳滨对虾和墨吉对虾制备简单eDNA 混合模板产生的引物偏倚程度影响较小,符合最初的实验设计预期。此时,2 个物种的模板浓度相同,高通量测序结果的偏倚主要是由于引物对不同模板的“偏好”程度不同产生的。在后续eDNA 宏条形码技术的数量性研究探索中,设计实验时可选用近源种作为实验对象,从而降低扩增过程中引物对不同模板偏好性不同而导致的“偏倚”现象。
3.2 eDNA 组成与高通量测序reads 数的相关性
当面对实际环境样本时,如果直接应用eDNA 宏条形码技术分析高通量测序reads 数与待测样本生物量的关系,从生物到eDNA 以及从eDNA 再到高通量测序结果的两个过程中的数学关系则难以理清。鉴于此,本研究采取“化繁为简”的策略,主要关注eDNA与高通量测序reads 数之间的关系。同时,考虑到复合底物的PCR 过程比较复杂(Harperet al, 2019),最终注释到每个物种的测序reads 数都受其反应体系的影响,故直接比较样本间的reads 数会产生一定偏差。充分考虑这些影响因素,本研究采用了样本内的2 个物种reads 数比值,用于确定其与模板eDNA 浓度的相关性。
统计分析实验组的reads 平均数比值与PCR 模板eDNA 比值2 组数据,进行一元线性回归分析,最终得出eDNA 组成与高通量测序reads 数的函数关系:y= 0.0716x+ 0.7043 (r2=0.9824)。等式中的斜率可反应出PCR 扩增过程中的偏倚程度(主要是引物偏倚),当扩增循环数相同,斜率越小,扩增过程中偏倚程度越大。等式中的截距主要是实验过程中的误差或者污染导致的。该等式可描述为:当环境水样中只存在墨吉对虾和凡纳滨对虾的DNA 时,可通过高通量测序的reads 数推算出2 个物种的相对生物量。
在实际操作中,可能由于模板eDNA 的回收率存在差异,使之不能完全符合预设的比例;另外,PCR扩增时并不是全程以底数为2 的指数式增长,且存在其他不可避免的系统误差,这些因素都会导致上述线性关系式出现一定偏离。但实验结果已充分表明,高通量测序reads 数与初始模板eDNA 含量呈线性正相关。
