PI3K 抑制剂联合地塞米松改善氧化应激细胞模型对激素的敏感性及其分子机制①
2021-08-23曾瑜真杜开锋金建军闵智慧毛若琳陈智鸿
曾瑜真 杜开锋 金建军 闵智慧 毛若琳 陈智鸿
(复旦大学附属中山医院呼吸与危重症医学科,上海200032)
糖皮质激素(glucocorticoids,GC)是治疗哮喘、慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)等慢性气道炎症最常用的药物,尤其在控制轻中度哮喘中效果最佳,然而重症哮喘、吸烟哮喘患者以及部分COPD 患者对激素治疗效果却不理想[1-2]。尽管重症哮喘占比较少,但其对GC 敏感性降低,由此导致的病死率较高,据报道每年因哮喘死亡的人数高达 25 万[3-4]。在 COPD 管理中,越来越多的研究提示目前推荐的高剂量ICS 获益不足[5]。因此,针对慢性气道炎症疾病开发更为有效的治疗方案,对于减轻患者和社会经济负担尤为重要。
氧化应激是哮喘、COPD 等慢性气道炎症的主要发病机制之一,它可使组蛋白去乙酰化酶(his‑tone deacetylase,HDAC)-2 活力降低、转录因子过表达和促炎信号通路增加等,这些可能导致激素不敏感[5-6]。氧化应激同时激活 PI3Kδ/Akt 信号通路,PI3Kδ 作为潜在的抗炎靶点可引起中性粒细胞活化,与哮喘和COPD对激素不敏感密切相关[7-8]。IL-8在中性粒细胞趋化过程中起关键作用,是COPD、哮喘等慢性气道炎症的促炎标志物,同时也可能是预测疾病严重程度的标志[9-10]。研究发现COPD 患者支气管上皮IL-8 表达的基线水平较高,可诱导黏蛋白基因表达导致气道黏液高分泌[11]。重症哮喘患者诱导痰上清中IL-8 浓度高于轻度哮喘患者,预示其炎症控制较差,预后不佳[3]。
本研究通过H2O2刺激人巨噬细胞系U937 细胞建立H2O2-TNFα-U937 细胞模型,模拟重症哮喘、吸烟哮喘患者外周血单个核细胞(PBMCs)的氧化应激-激素耐受效应。基于 IL-8 需在 TNF-α、LPS 等诱导剂作用下合成释放,本研究选取TNF-α 作为炎症诱导剂,单用PI3K抑制剂或PI3K抑制剂联合Dex干预细胞模型,探讨PI3K通路在慢性气道炎症中的作用机制,为临床治疗慢性气道炎症开辟新思路。
1 材料与方法
1.1 材料
1.1.1 实验细胞 人巨噬细胞系U937由中科院上海细胞生物学研究所提供。
1.1.2 主要试剂 Dex、TNF-α、H2O2购自美国Sigma公司;PI3K 抑制剂 BZE235、LY294002 购自 Selleck公司;IL-8 ELISA 试剂盒购自 R&D 公司;NF-κB、Jun、FOS 磷酸化抗体购自 CST 公司;TRIzol 购自Invitrogen 公司;荧光定量PCR 试剂盒及逆转录试剂盒购自 TaKaRa 公司;HDAC2 Assay Kit 购自 Abnova公司。
1.2 方法
1.2.1 模型建立与分组方案 ①H2O2-TNFα-U937细胞模型建立:U937 细胞分为未处理组与H2O2(400 µmol/L)刺激组,培养 4 h 后,分别加入0.1 µmol/L Dex 作用 45 min,各孔给予 20 ng/ml TNF-α 培养24 h,取上清,按照说明书采用ELISA 检测IL-8 的表达。各组设3 个复孔。②运用H2O2-TNFα-U937细胞模型筛选小分子抑制剂:U937细胞经400 µmol/L H2O2刺激4 h 后,分别与0.1 µmol/L Dex、1 µmol/L BEZ235/LY294002 单独作用45 min,各孔给予20 ng/ml TNF-α继续培养24 h,取上清,按照说明书采用ELISA 检测IL-8 的表达。各组设3 个复孔。③小分子抑制剂BEZ235/LY294002 联合Dex:U937 细胞经400 µmol/L H2O2刺激4 h 后,分别与 0.1 µmol/L Dex、1 µmol/L BEZ235/1 µmol/L LY294002 联合作用45 min,各孔给予20 ng/ml TNF-α 继续培养24 h,取上清,提取细胞RNA,ELI‑SA 及实时荧光定量PCR(Real-time PCR)检测各组IL-8 表达;提取细胞核蛋白和总蛋白,检测核蛋白HDAC2活力及NF-κB、AP-1蛋白磷酸化水平。各组设3个复孔。
1.2.2 Real-time PCR 检测各组细胞 IL-8 mRNA 表达水平 用TRIzol 法提取各组细胞RNA,按照TaKaRa 逆转录试剂盒说明书将RNA 逆转录为cDNA,按照PCR 试剂盒说明书进行操作,最后计算各组细胞IL-8 mRNA 表达水平。所需引物序列如下:IL-8:(F)5'-TTGCCAAGGAGTGCTAAAGAA-3',(R)5'-GCCCTCTTCAAAAACTTCTCC-3';GAPDH:(F)5'-CCACCCATGGCAAATTCCATGGCA-3',(R)5'-TCTACACGGCAGGTCAGGTCCACC-3'。
1.2.3 HDAC2活力的检测 用碧云天核蛋白提取试剂盒提取细胞核蛋白,然后按照HDAC2 Assay Kit说明书进行操作。
1.2.4 Western blot 检测各组细胞 NF-κB、AP-1 蛋白表达水平 以RIPA 裂解细胞提取总蛋白,BCA法测定蛋白浓度以保证蛋白质量相同。SDS-PAGE电泳后,65 V 恒压电转蛋白至 PVDF 膜,BSA 封闭,滴加Jun、FOS 磷素化一抗(1∶1 000)、NF-κB 磷酸化一抗(1∶1 000)、GAPDH 一抗(1∶1 000)4℃ 孵育过夜,TBS 缓冲液漂洗后加HRP 二抗,室温孵育1 h 后发光系统进行检测。采用Quantity One 软件进行相对定量分析。
1.3 统计学处理 采用SPSS13.0统计软件进行统计学分析,所得数据结果以表示。两组间比较采用Studentt检验,多组间比较用单因素方差分析(ANOVA),P<0.05 表示差异有统计学意义。使用GraphPad Prisn7软件作图。
2 结果
2.1 Dex 对 H2O2-TNFα-U937 细胞的抑制作用减轻 TNF-α 可刺激 U937 细胞分泌 IL-8,H2O2+TNF-α联合刺激,使得U937 分泌IL-8 的水平更高,为421.8 pg/ml。激素(Dex)可以抑制 TNF-α-U937 模型和H2O2-TNFα-U937模型中IL-8的分泌,但它使前者IL-8 分泌水平下降为73%,而使后者IL-8 分泌水平下降仅50%(图1)。该结果提示H2O2是氧化应激的产物,是活性氧(reactive oxygen species,ROS)成分的一种,细胞模型中添加H2O2可明显增加细胞释放炎症介质的能力,并使细胞对Dex 的抑炎作用产生抵抗(图1)。

图1 TNF-α 刺激 Dex 预处理的 H2O2-TNFα-U937 细胞时IL-8的表达Fig.1 Effects of TNF-α on IL-8 expression in H2O2-TNFα-U937 cell pre-incubated with Dex
2.2 单用PI3K 抑制剂不能明显抑制H2O2-TNFα-U937 细胞释放IL-8 由上述实验可知H2O2-TNFα-U937 细胞模型对Dex 的敏感性下降,有效模拟了重症哮喘、吸烟哮喘患者PBMCs 的氧化应激-激素耐受效应。为了了解PI3K 抑制剂能否抑制H2O2-TNFα-U937 细胞释放 IL-8,将 Dex、PI3K 抑制剂BEZ235 或LY294002 分别与其单独作用,检测各组在TNF-α 刺激后的IL-8 表达水平(图2)。结果显示单用 Dex 可减少 H2O2-TNFα-U937 细胞释放 IL-8 水平约 55%,而 PI3K 抑制剂如 BEZ235 或 LY294002 单独作用于H2O2-TNFα-U937 细胞仅仅使得IL-8 的释放减少23%、21%。说明单用PI3K 抑制剂不能明显抑制H2O2-TNFα-U937 细胞释放IL-8,不如单用Dex的抑制效果。
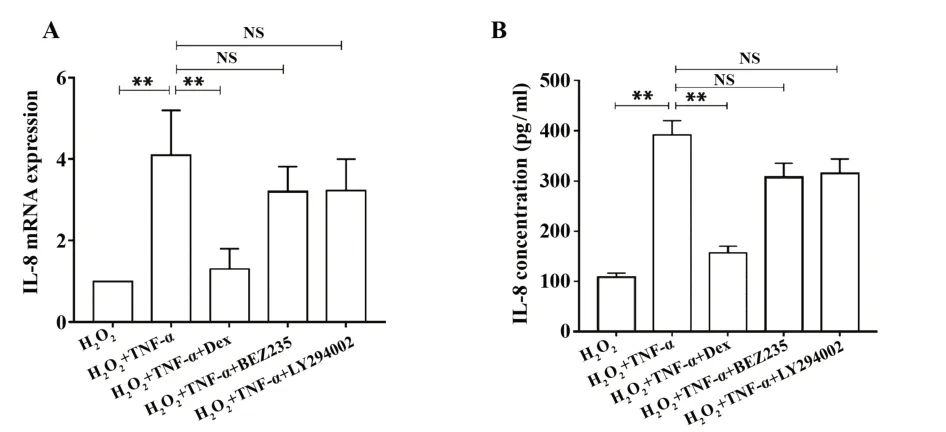
图2 PI3K 抑制剂干预后 H2O2-TNFα-U937 细胞的 IL-8 释放情况Fig.2 Effects of PI3K inhibitor on IL-8 releasing in H2O2-TNFα-U937 cell
2.3 PI3K 抑制剂联合Dex 明显改善H2O2-TNFα-U937 细胞模型对激素的不敏感性 为了进一步验证 PI3K 抑制剂联合 Dex 对 H2O2-TNFα-U937 细胞模型 的 作 用 ,将 Dex、PI3K 抑 制 剂 BEZ235+Dex 或LY294002+Dex 分别处理 H2O2-TNFα-U937 细胞,检测各组IL-8 分泌情况(图3)。组间比较显示BEZ235 或 LY294002 联合 Dex 对 IL-8 释放的抑制作用,对比Dex 单用抑制55%的情况下,又有明显降低。BEZ235+Dex 组、LY294002+Dex 组分别对 IL-8的分泌抑制为86%、65%(vs H2O2-TNF-α组)。

图3 PI3K 抑制剂联合 Dex 干预后 H2O2-TNFα-U937 细胞的IL-8释放情况Fig.3 Effects of PI3K inhibitor plus Dex on IL-8 releas⁃ing in H2O2-TNFα-U937 cell
2.4 BEZ235联合Dex可部分恢复H2O2-TNFα-U937细胞核蛋白HDAC2 活力 已知激素耐受与氧化应激损害了HDAC2 活力有关,为了验证PI3K 抑制剂BEZ235 或LY294002 辅助Dex 减轻该模型激素不敏感是否与HDAC2活力改善有关,提取上述各组细胞核蛋白检测其HDAC2 活力(图4)。结果显示H2O2-TNFα-U937 细胞模型组较空白对照组HDAC2 活力显著降低(P<0.01),说明氧化应激可损害U937 细胞核蛋白HDAC2 活力,在Dex 单独处理后能部分恢复其 HDAC2 活力,但不显著;BEZ235 联合 Dex 可进一步促进 HDAC2 活力恢复(vs H2O2组,P<0.05),Dex 联合LY294002 与H2O2组相比无明显差异。提示PI3K 的两个抑制剂对HDAC2 活力恢复能力不同。BEZ235 联合Dex 能更加有效地逆转该模型核蛋白HDAC2活力,从而改善其对激素的敏感性。

图4 Dex 或 PI3K 抑制剂联合 Dex 对 H2O2-TNFα-U937 细胞核蛋白HDAC2活力的影响Fig.4 Effect of DEX or PI3K inhibitor combined with Dex on activity of nuclear protein HDAC2 in H2O2-TNFα-U937 cell
2.5 PI3K 抑制剂(BEZ235/LY294002)联合Dex 可降低H2O2-TNFα-U937 细胞中炎症转录因子NF-κB、AP-1 的磷酸化水平 为了探索BEZ235/LY294002联合Dex 减轻H2O2-TNFα-U937 模型对激素不敏感的机制,通过 Western blot 检测各组 NF-κB 磷酸化水平及AP-1 亚基c-FOS 及c-Jun 蛋白磷酸化水平(图5)。结果表明H2O2处理使炎症转录因子(NF-κB、c-FOS、c-JUN)磷酸化水平显著高于空白对照组(P<0.01);Dex 单独处理后 NF-κB 磷酸化水平无明显变化,而 LY294002 联合 Dex 能显著抑制 NF-κB 磷酸化水平(P<0.01);BEZ235 或 LY294002 联合 Dex 均能显著抑制c-FOS 和c-JUN 磷酸化水平(P<0.01)。提示BEZ235 或LY294002 能通过减轻相应炎症转录因子磷酸化水平来辅助Dex发挥抗炎作用。

图5 PI3K 抑制剂联合 Dex 对 H2O2-TNFα-U937 细胞炎症转录因子磷酸化的影响Fig.5 Effects of PI3K inhibitor on phosphorylation of nuclear signal transcription factors in H2O2-TNFα-U937 cell
3 讨论
氧化应激是哮喘、COPD 等慢性气道炎症性疾病的主要发病机制之一,也是激素治疗不敏感的关键因素[2]。但激素不敏感的发生机制涉及多种途径,目前仍没有明确证据指明如何改善这一现象。本研究前期预实验分离了重症哮喘、轻中度哮喘和健康对照者的PBMCs,证实Dex 抑制重症哮喘组PBMCs 释放IL-8 的作用较后两组明显减弱(数据尚未发表)。因此,建立H2O2-TNFα-U937 细胞模型来模拟氧化应激-激素耐受效应,以求深入研究氧化应激对慢性气道炎症激素不敏感的作用并探讨改善途径。结果发现H2O2处理U937 细胞后,细胞释放IL-8 明显增多,对Dex 的抑炎作用产生了抵抗,提示H2O2引起的氧化应激的确能损害细胞对激素的敏感性。
烟雾暴露哮喘小鼠模型表明,氧化应激可激活PI3K 通路,促进炎症反应,从而降低激素敏感性,且PI3Kδ 和γ 是潜在的抗炎靶点[12]。因此本研究选取了 BEZ235 和 LY294002 两种 PI3K 抑制剂对 H2O2-TNFα-U937 细胞模型进行单独干预,结果表明单用PI3K 抑制剂不能明显抑制细胞释放IL-8,甚至不如单用Dex。显然单独抑制PI3K 通路并不能有效抑制炎症反应,这可能与抑制IL-8 基因表达还需通过GC-糖皮质激素受体(GR)复合物与IL-8基因相互作用实现有关,目前尚无证据能予以解释。本研究进一步采用上述两种PI3K 抑制剂联合Dex 对细胞模型进行干预,发现两种PI3K 抑制剂联合Dex 能明显改善细胞对激素的抵抗,与单用Dex 或PI3K 抑制剂相比有显著差异。联合Dex干预有效或许在一定程度上与本研究的假设符合,但具体原因需要更多实验进行探索。
组蛋白去乙酰化酶(HDACs)家族是一类细胞内酶,可催化组蛋白赖氨酸去乙酰化来调节基因表达,HDAC2 几乎仅存在于细胞核中,是关闭活化的炎症基因的关键核酶,因此,HDAC2 减少将引起炎症过程放大[13]。研究表明氧化应激能通过激活PI3K/AKT/mTOR 通路导致HDAC2 活力降低,与哮喘和 COPD 患者激素不敏感性的发生密切相关[7,14]。临床研究也发现肺内氧化应激水平高的重症哮喘患者HDAC2活力明显受损,而低浓度茶碱有望通过PI3Kδ 依赖机制将降低的HDAC2 活力恢复到正常水平,改善COPD、重症哮喘和吸烟哮喘患者对激素的不敏感[15]。为了验证小分子抑制剂辅助Dex改善细胞对激素的不敏感性是否与HDAC2 活力改变有关,本研究检测了细胞核蛋白HDAC2 活力,结果证实氧化应激损害了U937 细胞核蛋白HDAC2 活力,Dex 单独处理仅能部分恢复其HDAC2 活力,但并不显著,与上述研究中Dex 对炎症介质释放的抑制作用明显减弱一致;BEZ235 联合Dex 能进一步促进HDAC2 活力恢复,LY294002 联合 Dex 与 H2O2组相比差异无统计学意义,尽管该结果与LY294002 能改善激素抵抗的发现相矛盾,也许是由于LY294002作为非选择性抑制剂,干预了其他通路导致其恢复HDAC2的活力受到了影响。
GC 的间接基因调控中,GR 结合HDAC2脱去乙酰基,使转录因子如 NF-κB 和 AP-1 失去功能,抑制炎症基因转录[16],因此本研究检测了 BEZ235 或LY294002 联合 Dex 对 NF-κB、AP-1 磷酸化水平的影响,研究其改善激素不敏感性的机制。通过West‑ern blot 分析,发现 Dex 单独处理后 NF-κB 磷酸化水平无明显变化,BEZ235 或 LY294002 联合 Dex 均能显著抑制AP-1 磷酸化水平,但仅LY294002 联合Dex 能显著抑制 NF-κB 磷酸化水平,可见 BEZ235 或LY294002 联合Dex 改善激素不敏感性的调节机制复杂,在辅助Dex 发挥抗炎作用的不同环节中存在着一定差异。LY294002 联合Dex 虽不能恢复HDAC2 的活力,但能通过减少 NF-κB、AP-1 的磷酸化水平来辅助Dex 改善氧化应激导致的激素不敏感,这也说明激素不敏感的具体作用机制亟待后续的深入探索。
由于本研究细胞模型并非慢性气道炎症疾病患者的来源细胞,气道炎症环境又较为复杂,故在某些程度上存在局限性。但从一定意义上来说,它是模拟重症哮喘、吸烟哮喘患者PBMCs 对激素干预不敏感效应的体外细胞工具。通过该细胞模型,本研究证实PI3K 抑制剂联合Dex 可以通过恢复HDAC2活力和(或)抑制核信号转录因子磷酸化,在体外改善H2O2-TNFα-U937 细胞模型对激素的不敏感性。PI3K 抑制剂是否能辅助激素治疗因氧化应激导致的慢性气道炎症患者对激素的不敏感性还有待深入研究。
