稀土掺杂对LiFePO4性能影响的第一性原理研究*
2021-08-14钟淑琳仇家豪罗文崴吴木生
钟淑琳 仇家豪 罗文崴 吴木生
(江西师范大学物理与通信电子学院, 南昌 330022)
掺杂是提高LiFePO4体相电子电导率, 优化其电化学性能的重要方法之一.稀土元素因具有高的电子电荷、大的离子半径以及强的自极化能力, 成为掺杂改性的重要选择.本文利用基于密度泛函理论的第一性原理方法研究了稀土元素 (La, Ce, Pr) 掺杂的锂离子电池正极材料LiFePO4的性质.计算结果表明, 稀土元素掺杂均不同程度地增加了LiFePO4的晶格常数和晶胞体积.在脱锂过程中, 稀土掺杂后材料体积变化率明显减小, 材料的循环性能提升, 但电池能量密度下降.稀土掺杂使LiFePO4由原来的半导体特性转变为金属特性, 增加了材料的电子电导率.力学特性的计算表明稀土显著增加了LiFePO4材料的延展性.另外, La和Ce掺杂后的LiFePO4在Li离子迁移过程中表现出复杂的能垒变化, 在远离稀土离子处迁移势垒呈现出不同程度的减小, 而在靠近稀土离子处迁移势垒起伏较大.与Ce掺杂相比, La掺杂造成的离子迁移势垒的变化程度更大, 表明稀土离子掺杂对体系局域结构产生较大的影响.
1 引 言
锂离子电池由于具有高能量密度、高功率密度、高寿命、无污染等优点而广泛应用于便携式电子产品[1].近年来随着电动汽车和清洁能源储能需求的发展, 开发具有高能量密度、高功率密度、长循环寿命的锂离子电池十分必要.锂离子电池的性能在很大程度上取决于相关材料的性能, 特别是正极材料的综合性能.在商用的正极材料中, LiFePO4因具有理论容量高、成本低、环境友好等优点, 已被大规模应用于电动汽车、储能、备用电源等领域[2,3].然而, 受到固有的低电子电导率和离子电导率的影响, 随着充放电电流密度的增加, LiFePO4的实际容量会有明显的损失, 从而限制了其在锂离子电池上的进一步应用[4].为了提高LiFePO4的电导率, 人们不断地改善合成和加工策略.比如, 为了获得更好的导电性, Takahashi等[5]和Yamada等[6]把LiFePO4材料纳米化, 缩短扩散路径.Huang等[7]通过在LiFePO4的纳米复合材料中加入导电碳来提高其导电性能.然而, 这种方法并不能内在增强LiFePO4的体相电子电导率,并且通过添加炭黑降低了其储能密度.
通过进一步探索, 研究人员发现, 掺杂是提高LiFePO4体相电子电导率, 优化其电化学性能的重要方法之一.Chung等[8,9]和Shi等[10]通过对Li位进行Mg, Zr, Nb和Cr等高价金属阳离子掺杂,使LiFePO4的电子电导率提高了8个数量级, 并且Shi等[10]通过Li位的Cr掺杂还极大地降低了Li离子的活化能.除了Li位掺杂, Wang等[11]通过第一性原理的计算和实验, 证实了Fe位上的Mo掺杂也可以提高材料的电子电导率.以上研究表明, 掺杂可以有效地提高LiFePO4材料的导电性能.然而, 关于高价阳离子掺杂的物理机理一直存在争议, 比如Thackeray[12]认为LiFePO4电子电导率的提高不是掺杂高价金属阳离子的作用, 而是合成过程中残留碳的重大贡献.
除了高价金属阳离子掺杂外, 稀土元素由于具有电荷高、离子半径大、自极化能力强等特点[13],稀土掺杂被认为是改善锂离子电池正极材料性能的另一种有效方法.近年来, 有关锂离子电池正极材料稀土掺杂的研究越来越多, 比如, Ghosh等[14]通过在正极材料LiCoO2中掺杂稀土元素La, 提高了电极材料的电化学循环稳定性.Sun等[15]通过对稀土元素La, Ce, Nd和Sm掺杂后的LiMn2O4正极材料性能的研究, 证明了稀土掺杂能够稳定LiMn2O4的骨架结构, 并有效改善了LiMn2O4材料的电化学性能.Ding等[16]在三元材料LiNi1/3Co1/3Mn1/3O2中掺杂稀土元素La, Ce和Pr得到Li[Ni1/3Co1/3Mn1/3]1—xRExO2(RE = La, Ce, Pr)体系, 掺杂后材料的放电容量和循环性能都得到很大程度的提高, 同时电荷转移阻抗也得到了很好的压制.郑路敏等[17]利用第一性原理计算系统研究了La, Ce, Pr, Sm等稀土元素通过Mn位掺杂对Li2MnO3正极材料导电性的影响, 研究发现, 稀土元素掺杂对Li2MnO3的电子电导率均有不同程度的改善, 其中La元素最为显著.
就LiFePO4而言, 据我们所知, 稀土掺杂的研究报道不多, 且主要集中在Li位掺杂[18].比如,Luo等[19]通过在LiFePO4中的Li位掺杂La, 使得材料的可逆比容量和稳定性有了较大提高.进一步的研究发现, La在这里起到了减小颗粒尺寸、增加电导和Li离子迁移率的作用.关于LiFePO4的Fe位稀土掺杂, 尤其物理机理的研究, 目前仍鲜有报道.Fe位稀土掺杂是否像其他高价阳离子掺杂一样, 对LiFePO4材料的电导率会有所改善? 能否像LiCoO2中的Co位掺杂一样, 能有效地保护LiCoO2的结构稳定性, 抑制相变, 提高材料的循环性能[20]? 对这些问题的探讨能为LiFePO4导电性能的改善提供一种思路和方法.由于采用理论模拟的方法能够方便地预测材料的性能[21], 因此,本文采用第一性原理计算的方法研究稀土元素(La, Ce, Pr)在Fe位掺杂后LiFePO4的原子结构、脱锂电位和体积变化率、电子结构、力学性质以及离子迁移动力学性质, 从理论上预测稀土掺杂对LiFePO4的改性效果, 研究结果将为以后正极材料的设计、制备和优化提供必要的信息.
2 计算细节和方法
本文的所有计算都在基于密度泛函理论的VASP (Vienna ab initio simulation package)软件包中进行[22].原子实和价电子之间的相互作用通过缀加投影平面波方法来描述[23], 展开平面波的截断能设定为520 eV, 并采用了广义梯度近似(GGA)的Perdew-Wang (PW91)作为交换关联泛函[24].为了补偿计算中GGA方法对过渡金属元素Fe和稀土元素La强局域化的d轨道, Ce和Pr强局域化的f轨道所低估的关联效应, 我们施加了Anisimov等[25]提出的Hubbard修正(GGA +U)的方法来弛豫离子和晶胞.其中, Fe的有效U值取为3.7 eV[26], 对于稀土元素La, Ce和Pr, 有效U值分别取7.50 eV[27], 5.30 eV[28]和7.05 eV[29].掺杂结构采用1 × 2 × 1的LiFePO4超胞; 弛豫时布里渊区的数值积分采用Monkhorst-Pack方法[30], k点网格数为3 × 2 × 8; 计算电子结构时,网格数为6 × 6 × 12.因为体系具有磁性, 所以所有的计算都考虑自旋极化.对费米能级采用高斯展宽, 展开宽度取为0.2 eV.晶胞内所有原子进行了完全弛豫, 弛豫收敛精度为1 × 10—5eV/atom, 原子间的相互作用力不超过0.03 eV/Å.此外, 采用弹性能带(NEB)法[31]搜索Li离子的迁移路径,并计算迁移势垒.电子结构和力学性质的分析以及第一性原理轻推弹性带(FP-NEB)脚本的产生通过能源材料计算平台来完成[32].
3 稀土掺杂的LiFePO4的原子结构
首先, 根据实验结果[33], 构建空间群为Pnma的正交橄榄石结构的LiFePO4原胞, 每个原胞包含28个原子, 其中4个Li, 4个Fe, 4个P和16个O.Li和Fe位于八面体中心, P位于四面体中心,O原子以扭曲的六方密堆积方式构成晶胞的基本骨架.每个FeO6八面体由共同的顶点连接, 同时一个FeO6八面体与两个LiO6八面体和一个PO4四面体共边.另外, 根据Shi等[10]的第一性原理计算结果, 对反铁磁构型的LiFePO4进行优化, 优化后的晶格参数列于表1中, 其中a = 10.423 Å,2b = 12.139 Å, c = 4.753 Å.由表1可见, 本文的计算结果与实验值[33]和Shi等[10]的理论计算结果一致, 表明本文的计算方法是可靠的.
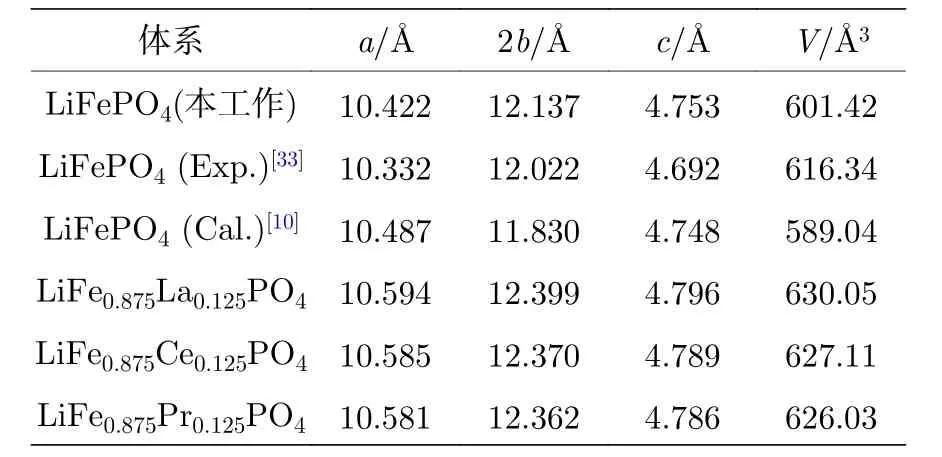
表1 未掺杂与稀土掺杂的LiFePO4的晶格常数和超胞体积Table 1.Optimized lattice contests and cell volume of LiFePO4 without doping and with rare-earth doping.
在获得了LiFePO4原胞的基态结构后, 利用1 × 2 × 1的LiFePO4超胞来构建掺杂结构, 将超胞中的1个Fe原子替换为稀土原子(La, Ce, Pr),如图1(a)所示.然后, 将掺杂后的结构进行优化,优化后的晶格参数也列于表1中.由表1可知,掺杂没有破坏原有的晶体结构, 但晶格常数明显增大, 同时晶胞的体积也增加, 显然这是因为稀土元素的离子半径比Fe离子大所导致的, 并且掺杂后晶格体积的变化与掺杂元素离子半径变化一致.

图1 (a) LiFePO4和(b) FePO4中稀土掺杂位置示意图Fig.1.Crystal structure of (a) LiFePO4 and (b) FePO4 with rare-earth doping sites.
为了进一步分析稀土掺杂对LiFePO4结构的影响, 计算了稀土掺杂后的LiFePO4结构中稀土离子与最近邻O离子之间的键长, 如表2所列.为了更好地进行比较, 表2还列出了未掺杂LiFePO4中Fe—O键长.由表2可知, 在未掺杂时, Fe—O键长分别为2.225, 2.080, 2.132和2.268 Å.当Fe位被稀土离子(RE)替代后, RE—O之间的键长都明显要大于掺杂前的Fe—O键长, 并且La—O,Ce—O和Pr—O的键长各不相同, 三者的平均键长分别为2.467, 2.401, 2.387 Å, 这种差异主要是由三种稀土离子的半径不一致引起的.在六配位的结构中, La离子的半径(1.03 Å)最大, Pr离子的半径(0.99 Å)最小, Ce离子半径(1.01 Å)介于二者之间, 所以导致La—O的平均键长最大, 而Pr—O的相对最小.相应地, 稀土离子掺杂对邻近Fe—O键长也有一定的影响, 与掺杂前相比, 稀土离子附近的Fe—O平均键长都略有增加.由此可见, 当LiFePO4中掺入稀土元素后, 在稀土离子的作用下, 掺杂结构的晶格常数都有所增加, 从而导致晶体体积也有所扩大.

表2 未掺杂LiFePO4中的Fe—O键长和稀土掺杂结构中RE—O键长Table 2.Bond lengths between Fe atoms and O atoms, rare-earth atoms and O atoms in LiFePO4 without doping and with rare-earth doping structure,respectively.
4 体积变化率与脱锂电位
电极材料脱嵌锂过程中体积变化率是一个影响电池循环性能的重要因素.分别计算了稀土掺杂后脱锂过程中的体积变化率 Δ V/V0, 其中V0代表未脱锂的LiFePO4或LiFe0.875RE0.125PO4(RE =La, Ce, Pr)晶胞的体积, Δ V 表示V0与完全脱锂后晶胞的体积之差.为了计算完全脱锂后材料的晶胞体积, 优化了完全脱锂态对应的FePO4的晶胞参数, 其晶格常数分别为a = 9.924 Å, 2b =11.767 Å, c = 4.867 Å, 与理论[34]和实验值[35]都符合得较好.在此基础上, 与LiFe0.875RE0.125PO4(RE = La, Ce, Pr)类似, 构建稀土掺杂后的Fe0.875RE0.125PO4(RE = La, Ce, Pr)结构, 如图1(b)所示.接下来分别优化各结构参数, 并计算各体系的体积变化率, 计算结果如图2(a)所示.由图2(a)可看出, 稀土掺杂后体积变化率都明显小于未掺杂体系, 由此可见, 稀土掺杂均在不同程度上抑制了材料在脱锂过程中的体积变化, 预示有利于材料循环性能的提升.此外, 从图2(a)可以进一步发现,分别通过La, Ce和Pr掺杂后的LiFePO4的体积变化率依次下降, 这种变化的产生可能是由La, Ce和Pr的离子半径和RE—O键长的不同引起的.由表2可知, RE—O键长的关系是La—O > Ce—O >Pr—O, 表明三种稀土离子掺杂中, Pr—O键最强,而La—O键最弱.因此, 在REO6八面体中, 三者的稳定性关系为LaO6< CeO6< PrO6, 由此可能造成脱Li前后La掺杂的LiFePO4体积变化最大,Pr相对最小, 而Ce介于二者之间.
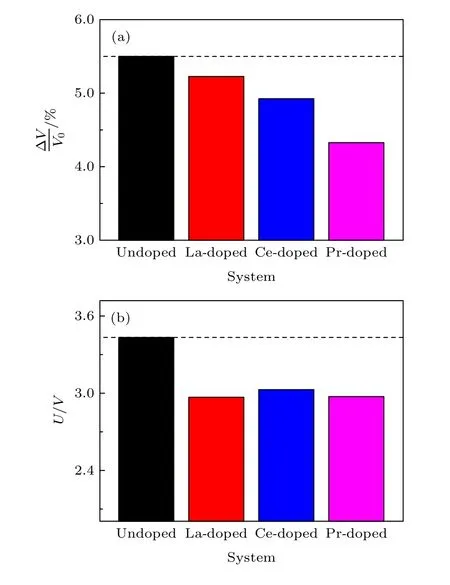
图2 LiFePO4 (a)完全脱锂后的体积变化率与(b)平均脱锂电位Fig.2.(a) Volume variations and (b) intercalation potentials of LiFePO4.
为了进一步解释稀土掺杂前后体系体积变化率下降的原因, 分析了Fe与配体氧之间的键长以及稀土元素与配体氧之间的键长在体系脱锂过程中的变化.未掺杂时, 在脱锂过程中, Fe—O键长有一个明显的变短过程, 其差值为0.118 Å; 而对于掺杂后的体系, 在脱锂过程中, RE—O(RE =La, Ce, Pr)键的键长变化并不大, 差值在0.049—0.068 Å范围内.由此表明, 掺杂前FeO6八面体的结构变化较大, 这种结构变化并不利于抑制材料在脱锂过程中的体积变化, 而掺杂后, 由于RE—O键比较稳定, 所以能一定程度地抑制材料体积的变化, 从而稳定材料的主体结构, 这将有利于材料循环性能的提高.
接下来研究稀土掺杂对LiFePO4材料平均脱锂电压的影响.对于未掺杂的LiFePO4, 平均脱锂电位的计算公式为
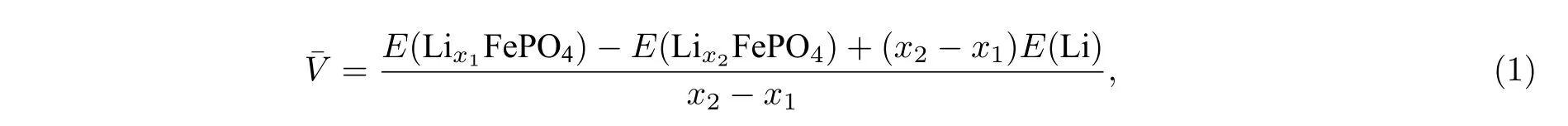
而对于稀土掺杂后的LiFePO4, (1)式可改为

其 中, E (Lix1FePO4) 和 E (Lix1Fe0.875RE0.125PO4) 为体系脱锂态的能量, E (Lix2FePO4) 和E(Lix2Fe0.875RE0.125PO4) 对应体系非脱锂态的能量, E (Li) 为一个bcc (体心立方)金属锂的能量, ( x2-x1) 为脱锂个数.此处计算 x1=0,x2=1 的平均电位, 计算结果如图2(b)所示.LiFePO4的平均脱锂电位为3.44 V, 与实验值一致[36].不过, 令人遗憾的是, 稀土掺杂后的脱锂电位大约为3 V, 比未掺杂的电位下降了近0.44 V左右, 意味着掺杂后正极的能量密度将有一定程度的下降.综上所述, 稀土掺杂使得LiFePO4正极材料在充放电过程中的体积变化率得到改善, 但不利于电池能量密度的提升.
5 稀土掺杂的LiFePO4的电子结构性质
图3(a)给出了未掺杂LiFePO4的自旋电子态密度图.可以看出, LiFePO4具有半导体属性, 带隙约为2.58 eV, 与文献值接近[37].另外, 计算发现在LiFePO4体系中, 所有的Fe均呈现+2价, 并都为高自旋构型, 磁矩为4μB.据晶体场理论, Fe2+离子中的6个电子占据Fe-3d轨道中的所有t2g和eg自旋向上轨道, 以及t2g的1个自旋向下轨道, eg自旋向下轨道为全空的状态.然而, 由于Fe在基态LiFePO4结构中以反铁磁的磁序排列, 所以体系中总的Fe2+自旋向上和自旋向下d轨道的态密度完全对称, 如图3(a)所示, 导致体系的总磁矩为0.La, Ce和Pr掺杂后, LiFePO4均呈现出金属性, 如图3(b)—(d)所示.掺杂前后LiFePO4电子结构的变化主要是由非等价离子替换造成的.为了进一步证实, 以La和Ce掺杂为例, 分析了掺杂后La和Ce的磁矩, 分别为0μB和1μB.La和Ce原子的最外层价电子排布分别为5d16s2和4f15d16s2, 价电子数目分别为3和4, 由此可知, 当La形成+3价离子时, 其最外层的3个价电子完全失去, 导致磁矩为0; 相似地, 当Ce形成+3价离子后还剩余一个f电子, 总磁矩为1.因此, 稀土离子在体系中多贡献1个电子, 使LiFe0.875RE0.125PO4(RE = La, Ce)形成金属性的电子结构.通过进一步计算发现, 稀土掺杂后LiFe0.875RE0.125PO4(RE = La, Ce)中每个Fe原子的磁矩仍然保持为4μB, 表明La和Ce原子上多失去的1个电子并没有转移到近邻的Fe2+上, 使Fe2+降价.因此, 在完美的体系中, 稀土离子多转移的电子并没有局域化.由此可见, 掺入稀土离子后, LiFePO4均由半导体属性转变为金属性, 从而改善了LiFePO4的电子电导率.
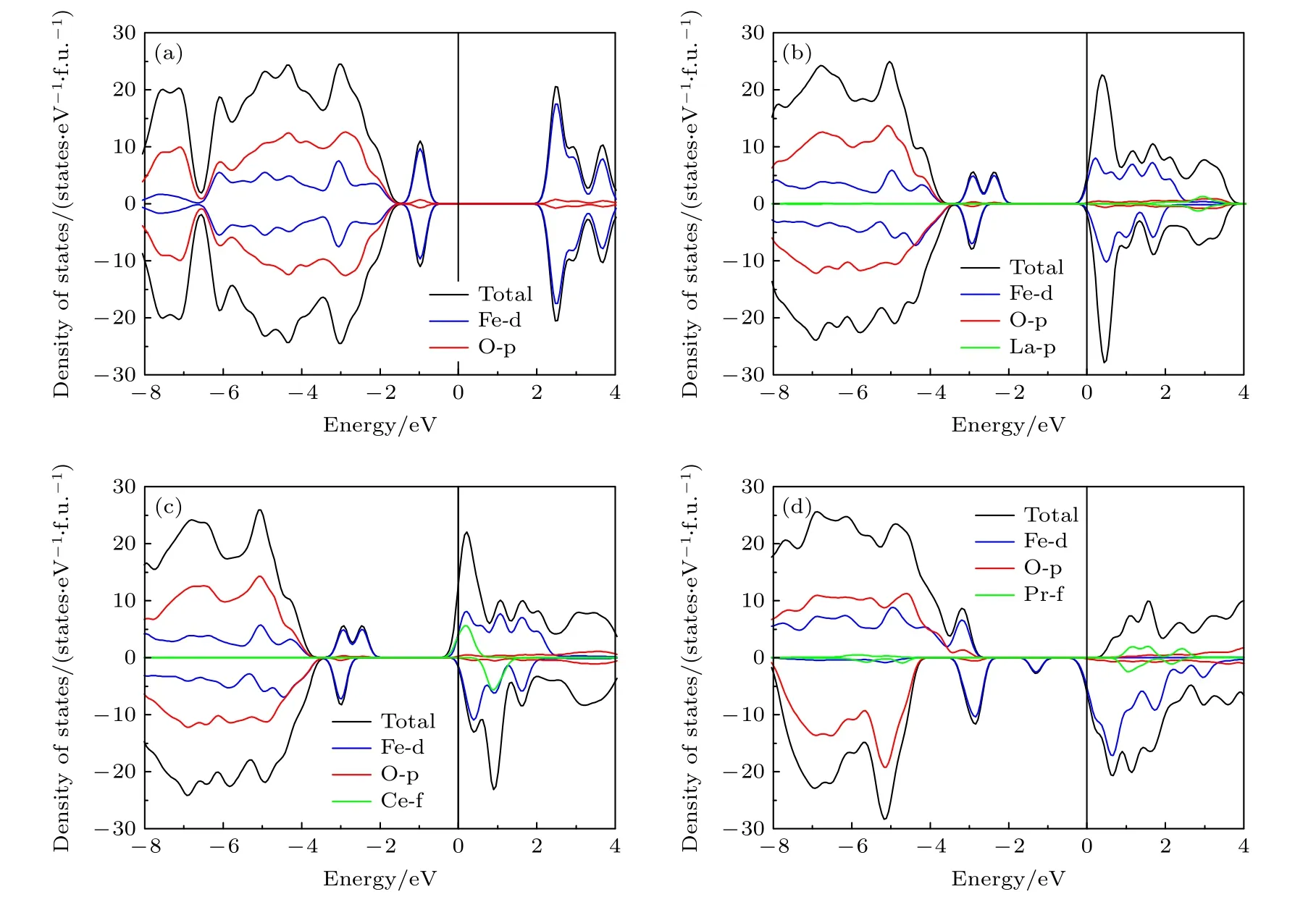
图3 LiFePO4的电子态密度图 (a)未掺杂; (b) La掺杂; (c) Ce掺杂; (d) Pr掺杂Fig.3.Density of states of LiFePO4: (a) Without doping; (b) La doping; (c) Ce doping; (d) Pr doping.
6 稀土掺杂的LiFePO4的力学性质
电池正极材料的机械稳定性在很大程度上影响其电化学性能.稳定性差会导致正极材料的相变和降解, 从而影响电池的充放电性能.因此可以预见, 充放电过程中机械稳定性与循环性能之间存在着重要的关系.表3列出了未掺杂的LiFePO4与La, Ce, Pr掺杂后的LiFePO4的弹性常数(Cij).由表3可知, 本文计算的LiFePO4弹性常数与文献中的值一致[38].

表3 LiFePO4未掺杂及稀土掺杂(RE = La, Ce, Pr)的弹性常数(单位: GPa)Table 3.Elastic constants (in GPa) of LiFePO4 without doping and with rare earth (RE = La, Ce, Pr) doping.
根据Born准则, 正交晶系的机械稳定性判据为[39]

根据表3列出的弹性常数可以计算, La, Ce, Pr掺杂前后LiFePO4均满足机械稳定性条件, 这表明稀土掺杂并未破坏材料的机械稳定性, 确保了掺杂的机械可行性.
在获得了各弹性常数后, 接下来计算掺杂前后LiFePO4材料的弹性模量.根据Voigt近似和Reuss近似, 对于正交晶系, 体模和剪切模量与弹性常数的关系可分别表示如下:

式中BV, BR和GV, GR分别表示Voigt和Reuss近似下的体积模量和剪切模量.Voigt和Reuss近似下分别获得的是材料弹性模量的上限和下限[40].对于多晶材料, 通过用Voigt-Reuss-Hill (VRH)方法按如下公式去估算材料的平均体积模量(B)和平均剪切模量(G):
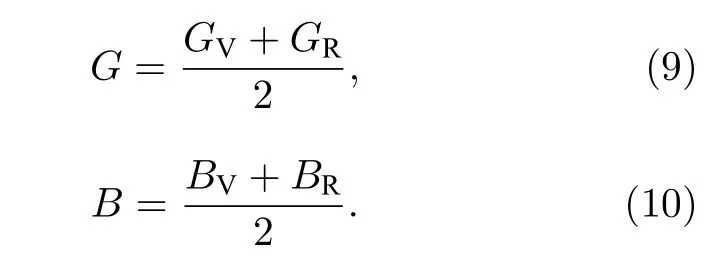
然后, 利用下面的关系式可以进一步确定杨氏模量(E)和泊松比(ν)[41]:
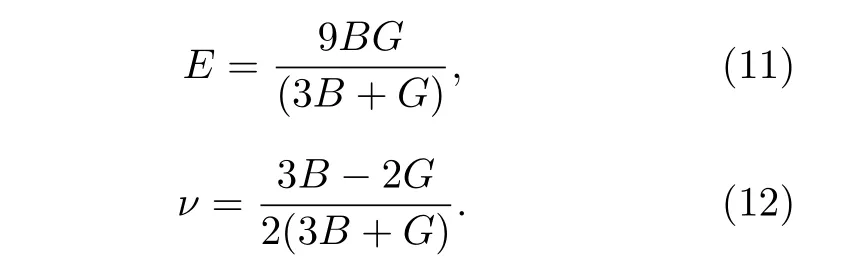
利用上述一系列关系, 分别计算了未掺杂的LiFePO4和La, Ce, Pr掺杂后的LiFePO4的体模量(B)、剪切模量(G)、B/G、杨氏模量(E)、泊松比(v), 计算结果列于表4中.对于LiFePO4, 本文的计算结果与其他的计算结果高度一致.体模B反映材料在弹性体系下对外界均一性压缩的抵抗能力; 剪切模量G是切应力与切应变的比值, 用来表征材料抵抗剪切应变的能力, G越大, 表示材料的刚性越强; 杨氏模量E是描述固体材料抵抗变形能力的物理量.由表4可知, La, Ce, Pr掺杂后, LiFePO4的B, G和E都明显变小, 表明稀土离子掺杂后, LiFePO4材料的强度和硬度都有不同程度的下降, 形变能力有所提高.泊松比(ν)是反映材料抵抗剪切形变的能力.泊松比越小, 材料在剪切形变的条件下就越容易保持稳定.La, Ce, Pr掺杂后的LiFePO4的泊松比有所增加, 表明相比于未掺杂的LiFePO4更容易发生剪切变形, 这与前面的结果一致.特别要提及的是, 作为锂离子电池材料, 材料的延展性对电池的循环性能和倍率性能有较大的影响.根据Pugh准则[42], 材料的延展性和脆性可通过B/G的大小来判断, 当B/G >1.75时, 认为材料具有较好的延展性, 否则材料是具有脆性的.从表4不难发现, 掺杂前, LiFePO4材料的B/G为1.92, 大于1.75, 说明LiFePO4材料具有较好的机械延展性; 当La, Ce, Pr掺杂后,LiFePO4的B/G值都有所增大, 分别为2.872, 2.15和3.00.由此可见, 相比于未掺杂的LiFePO4, La,Ce, Pr掺杂后, LiFePO4材料的延展性得到了进一步的提高.
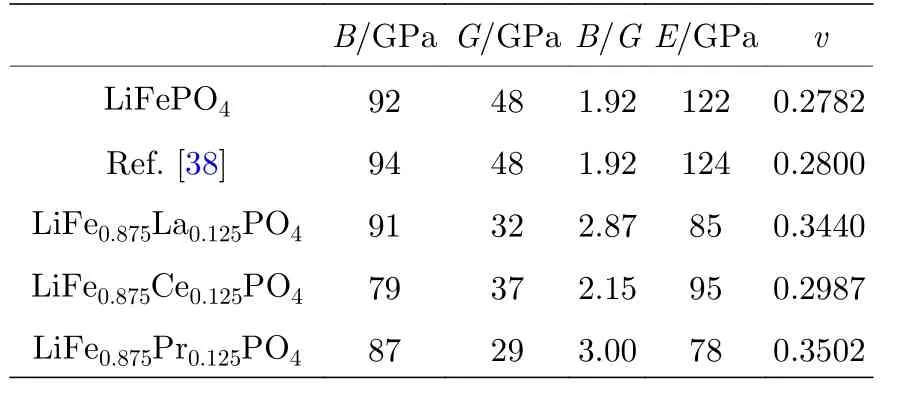
表4 未掺杂与稀土掺杂(RE = La, Ce, Pr)的LiFePO4的体模量(B)、剪切模量(G)、B/G、杨氏模量(E)、泊松比(v)Table 4.Bulk modulus (B), shear modulus (G),B/G, Young’s modulus (E), Poisson’s ratio (v) for LiFePO4 without doping and with rare earth (RE =La, Ce, Pr) doping.
7 稀土掺杂的LiFePO4中的Li离子迁移动力学
从所周知, Li离子在电极材料中的迁移能垒对锂离子电池倍率性能的影响至关重要[43].根据表1和表2, 由于Ce和Pr掺杂后的LiFePO4的晶格常数以及Ce—O和Pr—O键长都非常接近,说明Ce和Pr掺杂对LiFePO4结构产生的影响非常相似.因此, 为了简化分析过程和降低计算量,选取具有代表性的La和Ce元素掺杂的LiFePO4体系, 研究其中的Li离子迁移动力学.为了更全面地研究稀土离子掺杂对LiFePO4中Li离子迁移的影响, 选择在1 × 3 × 1的LiFePO4超胞中把其中1个Fe分别替换成稀土原子La或Ce (如图4所示), 此时掺杂浓度为1/12.考虑到实验和理论上已证实在LiFePO4化合物中, Li离子被约束在沿b方向的一维通道内迁移[44,45], 为简化起见, 本文只研究掺杂体系中Li离子在b方向的迁移通道.根据超胞中b轴迁移通道上Li离子与稀土离子的距离, 以及超胞中的周期性边界条件, 发现Li离子存在4条不同的迁移路径, 分别是1→2, 2→3, 3→4, 4→5, 而5→6与3→4是等价路径, 如图4所示.
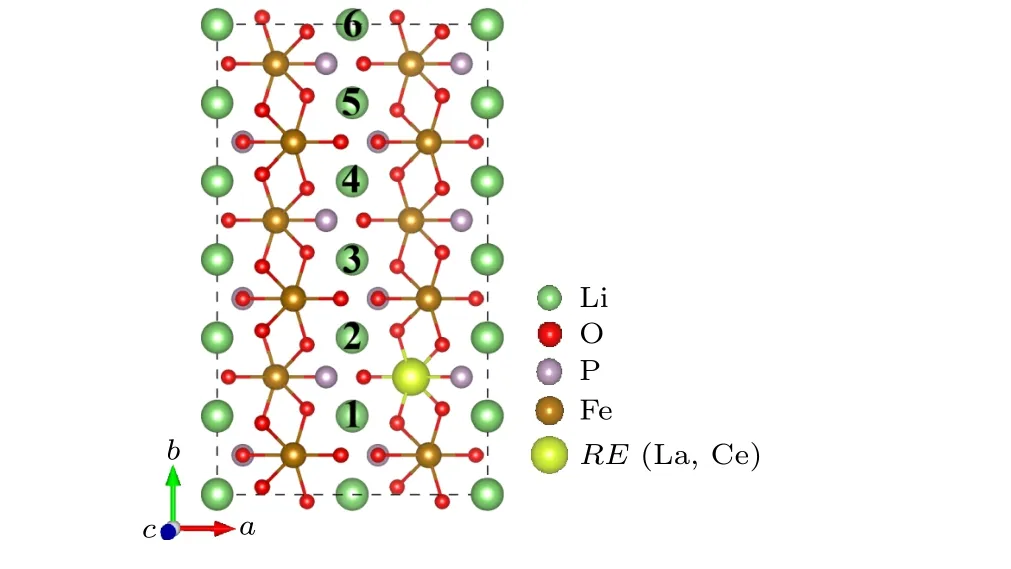
图4 稀土元素(La, Ce)掺杂后的LiFePO4中不同的Li离子迁移路径Fig.4.Different Li ion migration paths of LiFePO4 with rare-earth (La, Ce) doping.
为了比较稀土掺杂对LiFePO4中Li离子迁移的影响, 首先计算了Li离子在未掺杂的1 × 3 × 1的LiFePO4超胞中的迁移能垒.很明显, 在未掺杂的LiFePO4超胞中1→5包括的4条路径是完全等价的, 因此, 只计算了1→2路径上Li离子的迁移能垒, 为0.493 eV, 如图5所示, 比Ouyang等[45]的第一性原理计算结果小了0.1 eV左右.
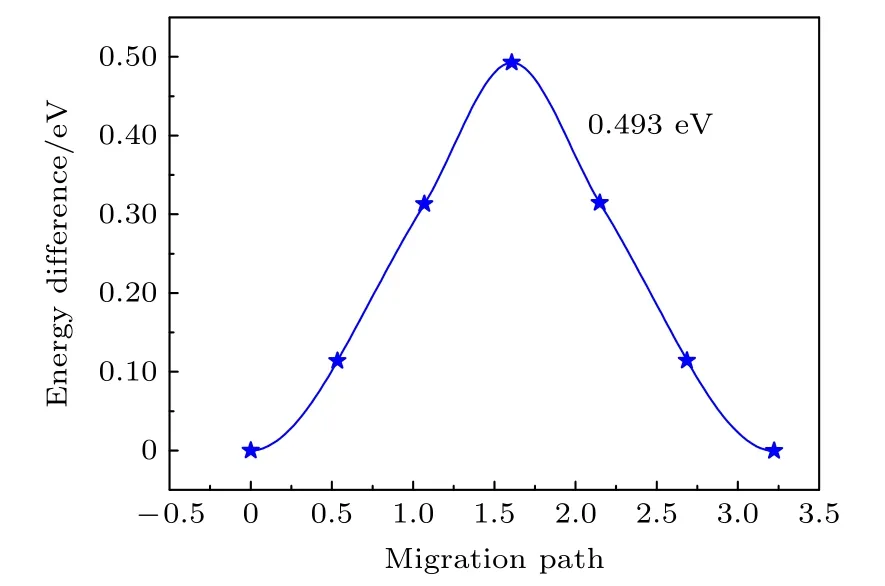
图5 未稀土掺杂的LiFePO4中Li离子迁移的能量分布Fig.5.Energy profile of the Li ion migration in LiFePO4 without rare-earth doping.
对于Li离子在La和Ce掺杂LiFePO4中的迁移, 分别计算了Li离子在上述4条路径中的迁移能垒, 如图6所示.根据图6所示的能垒, 发现Ce和La掺杂对LiFePO4中Li离子的迁移具有极为相似的影响.与未掺杂相比, 在La和Ce掺杂体系中, Li离子在靠近稀土离子时(1→2)的迁移能垒都非常大, 分别为1.429和1.255 eV, 是未掺杂的2倍多.我们推断如此大的能垒变化与稀土掺杂之后的局域结构变化大有关.以La掺杂为例,正如表2所列, La离子掺杂后, LaO6八面体中La—O平均键长为2.467 Å, 比未掺杂时的Fe—O键长(2.176 Å)增加了13%左右, 显然会导致La离子周围的迁移通道受到压缩, 从而增加Li离子在其中的迁移难度.对于Ce掺杂而言, Ce—O平均键长为2.401 Å, 要小于La—O键长, 表明Ce离子周围的迁移通道会略大于La离子周围, 所以导致Ce掺杂体系中1→2路径的能垒要小于La掺杂体系.与La和Ce近邻的Li离子迁移不同, 对于远离La和Ce离子处Li离子的迁移情况, 计算表明, 其迁移能垒范围分别为0.041—0.276 eV和0.061—0.454 eV, 如图6(a)和图6(b)所示, 与未掺杂体系中Li离子的迁移能垒相比, 有了明显的降低.这仍然是受到Li离子迁移通道大小的影响.根据表1所列, 与未掺杂相比, La和Ce掺杂后除了整个晶胞的体积都有所增大外, a方向的晶格常数也明显增加, 从而拉大了Li离子沿b方向的迁移空间, 减少了迁移能垒, 使得Li离子的迁移更容易发生.另外, 由于稀土离子掺杂对体系局域结构产生较大的影响, 使局域结构产生畸变, 由此引起势能面的较大波动, 从而导致Li离子的迁移能垒变化范围非常大.
从上面的计算结果可知: 一方面, 稀土掺杂整体上可以降低Li离子的迁移势垒, 提高迁移速率;另一方面, 稀土离子的掺入改变了局域结构, 使得周围的势能面起伏更大, 不利于Li离子的迁移.此外, 由图6的能垒数据可知, Li离子在1—5的完整迁移路径中, 对应的能量势垒呈现出明显的方向性, 即由靠近稀土离子的Li位(1或2号位)向远离稀土离子的Li位(3, 4和5号位)迁移时, 能垒较低, 而远端的Li向靠近离稀土离子的方向迁移时, 能垒极大.由此表明, 稀土掺杂后体系中Li离子倾向于向远离稀土离子的方向迁移, 在考虑实际迁移中, Li离子将会绕过稀土掺杂位而进行迁移.综上可知, 稀土离子的掺杂对Li离子的迁移有着重要的影响, 对于LiFePO4而言, 由于Li离子只有沿b方向的一维扩散通道, 如果Li离子通过稀土离子附近迁移的话, 将会严重影响材料的倍率性能, 因此, 在实际稀土掺杂时, 稀土离子的浓度需要控制在合理范围, 使Li离子尽可能向远离稀土离子的方向迁移.

图6 Li离子在La和Ce掺杂LiFePO4中的迁移路径和势垒 (a) La掺杂; (b) Ce掺杂Fig.6.Diffusion paths and energy barriers of Li ions in Laand Ce-doped LiFePO4: (a) La-doped;(b) Ce-doped.
8 结 论
本文采用第一性原理的方法研究了稀土(La,Ce, Pr) 掺杂的锂离子电池正极材料LiFePO4的结构、脱锂电位和体积变化率、电子结构、力学性质以及离子迁移动力学性质.结果表明, 我们所考虑的稀土元素掺杂均增加了LiFePO4的晶格常数和晶胞体积, 掺杂后晶格体积的变化与掺杂元素离子半径变化一致.在脱锂过程中, 稀土掺杂后材料体积变化率明显减小, 材料的循环性能将得到提升, 但能量密度下降.La, Ce, Pr掺杂使LiFePO4由原来的半导体特性转变为金属特性, 稀土掺杂提高了材料的电子电导率.稀土掺杂增加了LiFePO4材料的延展性.从La和Ce掺杂的LiFePO4中Li离子迁移的情况来看, 在远离稀土离子处迁移势垒呈现出不同程度的减小, 而在靠近稀土离子处迁移势垒起伏较大, 特别是在稀土离子最近邻处的Li离子迁移势垒明显增大.与Ce掺杂相比, La掺杂造成的离子迁移势垒的变化程度更大.
