不同前驱体制备硅/石墨相氮化碳材料用于锂离子电池*
2021-08-04陶俊岩刘慧添王晓一刘元生戴晓谦单忠强
陶俊岩,刘慧添,王晓一,刘 旭,刘元生,戴晓谦,单忠强
(天津大学 化工学院,天津 300350)
0 引 言
锂离子电池因其具有高循环稳定性、高能量密度、环保等特点,在新能源材料领域引起了广泛的关注。然而,目前商业化石墨负极由于较低的理论比容量(372 mAh/g)很难满足人们的需求。硅作为最有希望取代工业石墨负极的材料之一,具有储量丰富、环境友好等明显优势,其理论比容量高达4 200 mAh/g,与石墨负极相比具有更高的潜力[1]。但是在锂化/脱锂过程中,它带来300%的体积膨胀,会导致硅颗粒的粉碎和容量迅速衰减[2]。为了缓解硅体积膨胀的问题,将碳材料与纳米硅复合是一种有效的解决方式[3]。石墨相氮化碳(g-C3N4)具有叠层二维结构,每个平面片由sp2杂化共轭C和N原子组成,建立π-共轭电子结构[4]。 g-C3N4的主要合成方法有固相反应法、溶剂热法[5]、热缩聚法[6]、电化学沉积法。g-C3N4耐酸碱腐蚀,具有很好的机械强度[7],高热稳定性以及一定的导电性,目前在催化传感等一系列领域得到广泛应用[8-9]。近年来,研究者们报道了氮化碳的合成以及其作为载体材料在锂电池中的作用。Yao等[10]以尿素、三聚氰胺、硫脲、双氰胺为原料,在相同条件下通过直接聚合制备了石墨氮化碳(g-C3N4)材料,制备的U-CN(来自尿素)、M-CN(来自三聚氰胺)、T-CN(来自硫脲)和D-CN(来自双氰胺)展示了显著不同的微观结构,合成的g-C3N4粉体用作锂硫电池的硫基底,电化学性能表明,尿素衍生C3N4在500次循环后具有良好的循环稳定性。Wang等[11]采用无模板自组装和热处理的组合方法合成了具有三维多孔结构的Si@rGO/g-C3N4复合材料。然而,用不同的前聚体采用热缩聚法合成g-C3N4/Si复合负极材料的报道较少。热缩聚法是利用具有三嗪环结构的化合物为前驱体受热,高温诱导本体发生缩聚反应以生成g-C3N4。由于该方法对设备要求较低、操作简单、方便控制、可一步到位,本文以尿素、硫脲和三聚氰胺(价格低廉且氮碳比相同)为原料,通过热缩聚反应合成g-C3N4,再与硅复合形成负极材料。利用各种表征手段分析了前驱体对合成g-C3N4和g-C3N4/Si复合材料物理化学性质和电化学性能的影响,优化了g-C3N4/Si复合材料前聚体的选择。
1 实 验
1.1 材料与试剂
尿素(H2NCONH2,99 %),天津光复科技发展有限公司;硫脲(CH4N2S,99 %)和无水酒精,天津元立化工有限公司;三聚氰胺(C3N3(NH2)3,99 %),北京伊诺凯科技有限公司;纳米硅(直径50~80 nm),广州宏武材料科技有限公司,实验中的试剂均未经进一步提纯。
1.2 材料的制备
将6 g尿素研磨后放于石英舟,在管式炉中以2.5 ℃/min加热至550 ℃,并保温6 h。待降至室温后研磨,得到C3N4-u。称取0.08 g C3N4-u,加入到盛有30 mL无水乙醇的烧杯中,另取0.08 g Si,加入到盛有50 mL无水乙醇的烧杯中。超声分散1 h后,将混合悬浊液超声搅拌2 h,最后将混合溶液在80 ℃水浴蒸干,得到复合材料Si@C3N4-u。在相同条件下以硫脲和三聚氰胺为前聚体得到的产物分别表示为Si@C3N4-t和Si@C3N4-m。
1.3 材料的表征
本实验采用日本Rigaku公司的D/MAX-2500型X射线衍射仪来分析样品的晶体结构及物相组成。测试条件为Cu靶Kα射线,工作电压和电流分别为40 kV和200 mA,扫描范围为10~80°,扫速 8°/min。采用日本Hitachi公司的S-4800型扫描电子显微镜来观察复合材料的表面形貌。采用日本JEOL公司的JEM-2100F型透射电子显微镜来对材料进行微观结构分析。采用美国麦克公司生产的TriStarⅡ3020型分析仪进行测试,利用BET(Brunauer-Emmett-Teller)模型计算材料的比表面积,用BJH(Barret-Joyner-Halenda)模型计算材料的孔径分布。采用美国Perkin Elmer公司的Spectrum 100型红外光谱仪对材料中的官能团进行检测。采用德国NETZSCH公司生产的STA449F5型分析仪在氮气气氛下进行测试,测试温度为室温到800 ℃,升温速率为10 ℃/min。采用英国ThermoFisher Scientific公司生产的K-Alpha+型能谱仪进行测试。该仪器以Al Ka为X射线源,并以污染碳 C1s(284.8 eV为校正值)校准结合能,真空条件为5×10-8Pa。
1.4 材料电化学性能测试
以海藻酸钠(SA)为粘结剂,导电炭黑Super P为导电剂,将复合材料,海藻酸钠和Super P按照8∶1∶1的质量比放入玛瑙研钵,加入适量水研磨使浆料均匀分散。将混合后的浆料均匀涂覆在铜箔上,在鼓风干燥箱中55 ℃干燥10 min,然后在真空干燥箱中60 ℃下干燥8 h。最后将烘干的极片裁剪成直径为12 mm的圆片并作为工作电极,活性物质载量为1 mg/cm2左右。以金属锂片为对电极和参比电极,微孔PP/PE/PP复合膜为隔膜,1M LiPF6的EC/DEC溶液加入质量分数10%的氟代碳酸乙烯酯(FEC)为电解液,在氩气氛手套箱中组成CR2032型纽扣电池,静置12 h后进行电化学性能测试。
本实验采用武汉蓝电电子股份有限公司的CT2001A型电池测试系统对电池进行充放电测试,测试电压范围为0.01~1.5 V。通过测量得到的充放电循环性能和倍率性能曲线来判断电极的循环稳定性等性能。采用上海辰华CHI660E型电化学工作站对电池进行循环伏安(CV)测试,扫描电压范围为0.01~1.5 V(vs.Li+/Li),扫描速度为0.1 mV/s。
2 结果与讨论
2.1 材料的形貌与结构表征
图1 (a)为3种前驱体制备的纯g-C3N4材料的XRD图。由图可见,3种前驱体所制得的g-C3N4材料在13.1°和27.5°有明显的峰,该峰为石墨相结构的典型衍射峰,说明制备的g-C3N4具有与石墨相似的层状结构。13.1°处的峰归属于类石墨C3N4的(100)晶面,代表环状单元平面结构。27.4°处的峰归属于类石墨C3N4的(002)晶面,代表共轭芳香族片层的层间堆积[12]。三者的衍射峰强度有所不同,比较27.5°左右的衍射峰,由三聚氰胺和硫脲制成的g-C3N4的峰强于尿素制成g-C3N4的峰,说明C3N4-m及C3N4-t的结晶程度较好,而由尿素制成的g-C3N4片层少且厚度薄。图1 (b)为Si分别与3种材料复合得到的Si@C3N4XRD图。其中位于2θ=28.442°、47.302°、56.121°、69.130°、76.377°处的衍射峰分别对应着硅(PDF#27-1402)的(111)、(220)、(311)、(400)、(331)晶面[13]。衍射图谱没有发现其他杂峰,证明材料在制备过程中没有引入其他杂质。3种Si@C3N4复合材料在2θ=28°左侧均有一个较小的峰,说明其既包括了单质硅的特征峰,又包括了g-C3N4的特征峰。图1 (c)为3种原材料的红外光谱图。3种原材料的红外光谱图基本相同,3 500~3 000 cm-1区域的峰对应g-C3N4分子中N-H,O-H键的伸缩振动以及所吸附的H2O和NH3分子,1 200~1 700 cm-1处的吸收峰是由g-C3N4杂环上的C-N,C=N伸缩振动引起的。810 cm-1处的峰代表了g-C3N4三嗪环结构单元的弯曲振动[14],说明3种材料均为石墨相的C3N4。

图1 g-C3N4的XRD图以及Si@C3N4的XRD图和g-C3N4的红外图谱Fig 1 XRD patterns of g-C3N4 and Si@C3N4, and FT-IR of g-C3N4
图2(a)为C3N4-u的SEM 图,可以看出尿素煅烧后形成的C3N4-u呈花簇状多孔片状结构,片层厚度约为20 nm,且片层之间较松散。这是由于尿素作为前聚体进行热聚合反应中产生大量氨气,气体的搅动会导致氮化碳片与片之间的分离,增加了其比表面积[15]。图2(b)为C3N4-u与硅复合后的SEM图,硅堆积在C3N4-u形成的花簇结构中,但Si@C3N4-u整体结构较松散。如图2(c)所示,三聚氰胺煅烧后形成规整明显多层的C3N4-m,层间堆积较致密,部分结为平板块状。图2(d)为C3N4-m与硅复合后,硅堆积在C3N4-m表面,片层致密呈小块状。图2(e)为硫脲煅烧后形成C3N4-t的SEM图,C3N4-t同样为较为规整的片状多孔结构,片层厚且层与层之间间距较大。通过超声搅拌处理,图2(f)中可看出Si纳米粒子较均匀地分布在多孔片层的C3N4-t上,部分Si纳米颗粒夹嵌在C3N4-t片层之间以及进入大孔中,硅周围的C3N4-t不仅可以缓解Si的体积膨胀,减少结构塌陷对于电化学性能的影响,还可以减少团聚现象,有助于增大Si纳米颗粒与电解液的接触面积。

图2 g-C3N4以及Si@C3N4的SEM图Fig 2 SEM images of g-C3N4 and Si@C3N4
图3为3种氮化碳的TEM照片。如图3(a)C3N4-u呈花絮状分布,片层松散且孔较大,并伴有一定程度的片层粉碎。图3(c)C3N4-m则为显示为厚实的片层堆积,呈块状分布,图3(e)可以看出C3N4-t为大的卷曲片层,片层间距适中,大小厚度分布均匀。图3(b)为Si@C3N4-u的TEM图。可见Si稀疏分散在片层上与片层间,片层厚度薄,很难限制Si在充放电过程中的体积膨胀。Si@C3N4-m的TEM图3(d)可以看出片层堆积严重,大部分Si都团聚在氮化碳片层表面。而Si@C3N4-t的图像图3(f)可以看出Si能够进入氮化碳片层之间,片层厚度间距适中,这种夹层结构可以较好的抑制硅的体积膨胀,提高材料的电化学性能。

图3 g-C3N4以及Si@C3N4的TEM图Fig 3 TEM images of g-C3N4 and Si@C3N4
图4(a)、(b)为3种g-C3N4的BJH孔径分布曲线和氮气吸附脱附等温曲线,从图可以看出3种g-C3N4材料都具有典型的IV型等温吸附曲线,吸脱附曲线不重叠说明g-C3N4材料具有多孔结构[16]。通过分析可以得出,C3N4-u、C3N4-t、C3N4-m的 BET 比表面积分别为 50.0027,9.1290和 0.9565 m2/g。可以看出由尿素制备所得g-C3N4的BET比表面积最大,因为在尿素缩聚过程中产生大量CO2,NH3等气体,导致片层松散、破碎,由硫脲制备的g-C3N4比表面积适中,而由三聚氰胺制备的g-C3N4由于片层堆积较为致密,所以有着最小的比表面积。通过 BJH 进一步对材料的孔径进行分析,可知3种材料都存在着介孔,小孔径的孔有利于电解液的渗透和锂离子的传输。如图4(c)为3种原材料在氮气氛围下的TGA图像。由图可知三种g-C3N4原材料在300 ℃内热稳定性较好,没有明显的重量损失,300~500 ℃范围内,样品的质量损失很小,这是由于样品中吸附的水分子和挥发性组分的释放而导致部分失重。500 ℃以后,由于g-C3N4的分解,造成样品重量大量损失。C3N4-u、C3N4-t、C3N4-m分别在690,730,760 ℃分解完全,几乎没有残余物质剩留[17]。如图4(d)为Si@C3N4复合材料在氮气下的TGA分析图像。由图可知,当达到一定温度时,样品质量不再损失,这是氮化碳分解完之后剩余Si颗粒的质量。Si@C3N4-u、Si@C3N4-t、Si@C3N4-m分别残留了51.6%,52.3%,52.9%的Si,证实了复合材料中硅与氮化碳的比例基本都为1比1。
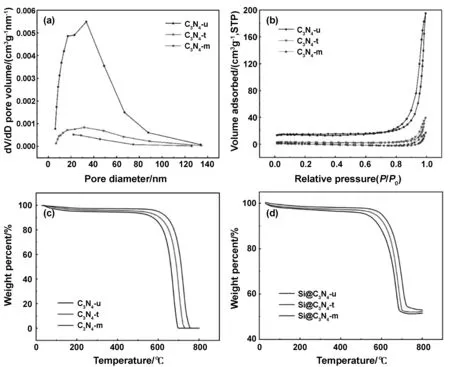
图4 g-C3N4的孔径图(a)、吸附脱附曲线图(b)以及在氮气下3种g-C3N4与Si@g-C3N4的热重图(c)、(d)Fig 4 Pore size distribution and nitrogen adsorption-desorption isotherms of g-C3N4 and thermogravimetric analysis results for g-C3N4 and Si@g-C3N4 under nitrogen atmosphere
图5(a)为3种原材料制备得C3N4-u,C3N4-m,C3N4-t的XPS全谱,可以看到C、N、O元素的存在,样品中O元素主要是由于聚合过程中吸收空气中的不定含氧物质(如水和二氧化碳等)引起的,这是一种氮化碳合成过程中比较常见的现象[18]。C3N4-u的C、N比为0.82,C3N4-t的C、N比为0.743,C3N4-m的C、N比为0.81,在误差范围内基本与理论C3N4的C、N比一致。图5(b)为3种复合材料的XPS全谱,可以看到C、N、O、Si元素的存在。图5(c)为C3N4-t材料的S 2p图谱,硫元素的含量仅有0.21%,163.8 eV处的峰代表了C-S键,168.649 eV处的峰对应了S-O键[19],说明了材料表面可能还残留着一些含S的基团。图中98 eV附近的峰对应于Si-Si,证明了零价单质Si的存在。102 eV附近的峰代表了Si-O键,这是由于小部分Si在空气中不可避免的氧化形成了SiO[20]。

图5 g-C3N4、Si@g-C3N4的XPS全谱(a)、(b)以及C3N4-t的S 2p高分辨图谱(c)和Si@g-C3N4的Si 2p高分辨图谱(d)Fig 5 XPS survey spectra of g-C3N4 and Si@g-C3N4, high-resolution S 2p spectra of C3N4-t and high-resolution Si 2p spectra of Si@g-C3N4
进一步对C 1s精细谱进行分峰操作,可以得到3个独立的峰如图6所示, 284.7 eV处的峰常被归因于C-CC=C或不定碳,286 eV附近的峰代表了C=N,,而288 eV附近的峰对应了C-N[21]。图7为3种原材料以及复合材料的N 1s精细谱。398.8,400.0和401 eV,附近的峰分别代表了吡啶N,吡咯N,石墨N。3种g-C3N4中,C3N4-u、C3N4-t、C3N4-m的吡啶N含量分别为61.2%、62.8%、59.3%。
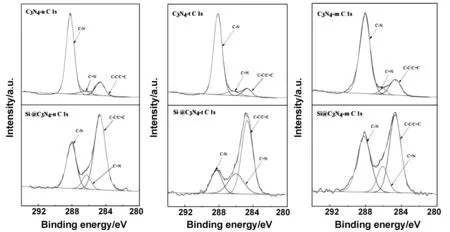
图6 g-C3N4、Si@g-C3N4的C 1s分峰高分辨图谱Fig 6 High-resolution C 1s spectra of g-C3N4 and Si@g-C3N4
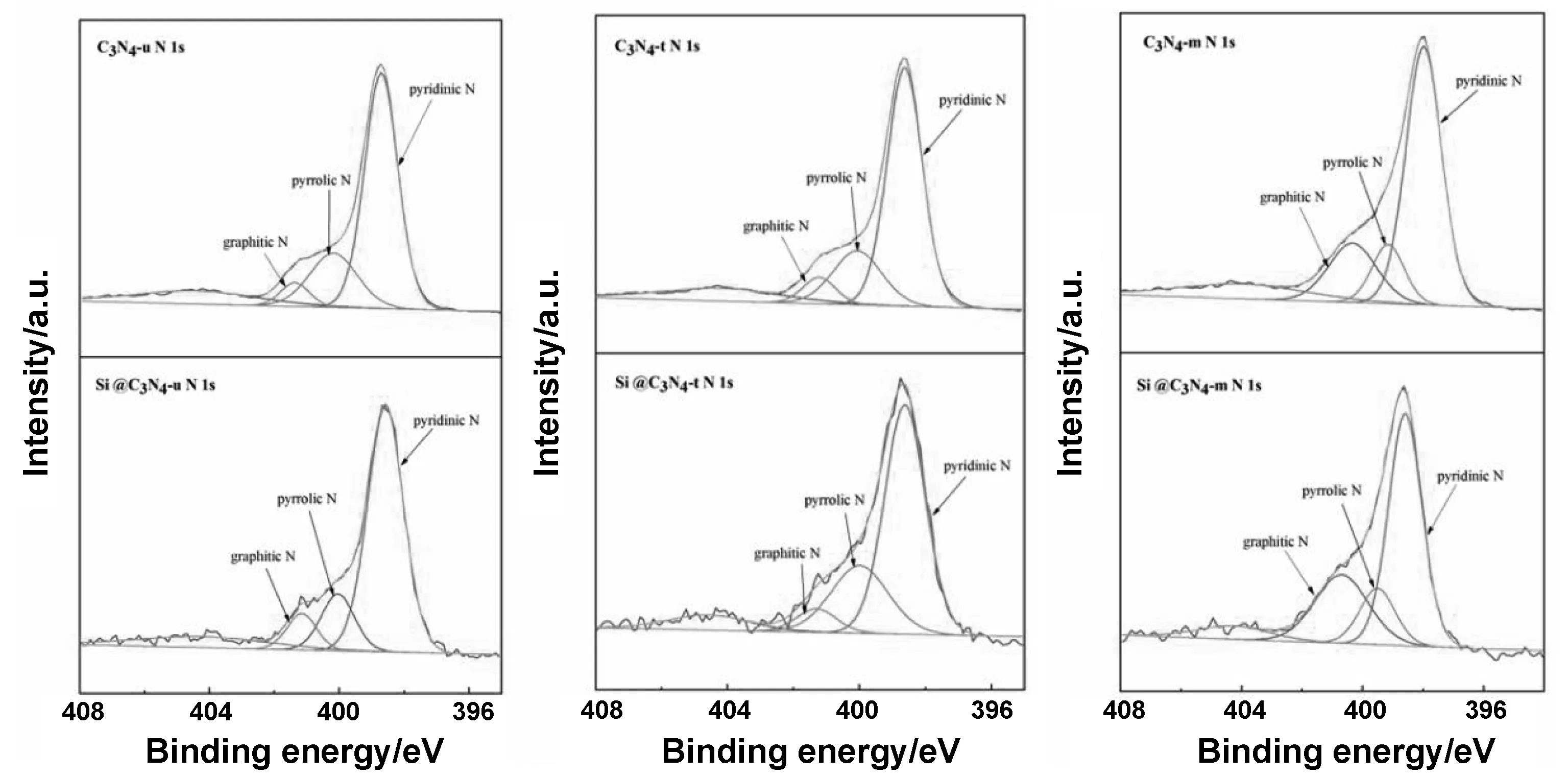
图7 g-C3N4、Si@g-C3N4的N 1s分峰图谱Fig 7 High-resolution N 1s spectra of g-C3N4 and Si@g-C3N4
为了验证3种氮化碳材料与硅的组装过程,将Si粉、C3N4-u与Si粉、C3N4-t与Si粉、C3N4-m与Si粉分别在反应体系中的浓度超声分散,静置15 h后观察分散液的沉降情况。图8给出了沉降结果,可以看出,C3N4-t/Si发生了明显的沉淀,上层液体澄清透明,这种现象可以归结于Si表面有一定程度的氧化而带有的-OH与C3N4-t材料表面残留的含S基团形成了氢键作用,进而引导硅沉积到C3N4-t的表面。

图8 Si粉、C3N4-u与Si粉、C3N4-t与Si粉、C3N4-m与Si粉的静置沉降实物图Fig 8 Static settlement physical diagram of Si powder, C3N4-u and Si powder, C3N4-t and Si powder, and C3N4-m and Si powder
2.2 材料的电化学性能
图9(a)、(b)、(c)分别为Si@C3N4-u、Si@C3N4-t、Si@C3N4-m复合材料1~5个循环的循环伏安特性曲线,扫描倍率0.0001 V/s,扫描范围0.01~1.5 V。在首次还原过程中,从1.1~0.7 V处出现了一个较宽的还原峰,归因于材料表面SEI膜的形成。在之后的循环中,在0.20和0.02 V处的两个阴极峰对应于锂离子插入电极材料,分别由于无定形Si向LixSi和无定形LixSi向结晶Li15Si4的相变[23]。在脱锂过程中,有两个相应的氧化峰分别出现在约0.30和0.50 V处,对应于晶体Li15Si4转变为无定形LixSi,而后转变为无定形Si的相变[24]。此外,CV曲线中阳极和阴极峰强度的增加表明复合材料在前5个循环中逐渐活化。Si@C3N4-t复合电极CV曲线在循环5圈后重合度较好,说明电极更稳定。3种复合材料中,Si@C3N4-t复合材料具有更高的峰值电流且峰面积较大,因而具有更高的嵌锂能力。
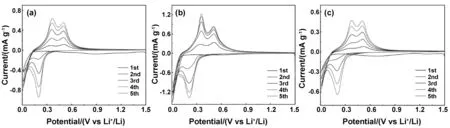
图9 Si@g-C3N4复合材料的CV曲线图Fig 9 Cyclic voltammetry profiles of Si@g-C3N4 compound material
如图10(a)为Si、Si@C3N4-u、Si@C3N4-m、Si@C3N4-t在200 mA/g的电流密度下活化三圈进而在500 mA/g的电流密度下进行的循环性能测试曲线。纯Si呈现出较高的初始充电比容量,但随后急剧衰减,这是由于Si纳米颗粒在充放电过程中巨大的体积效应导致电极粉化,使得活性物质表面的SEI膜不断破裂和生成,造成库伦效率低、循环性能差。以尿素作为前驱体的Si@C3N4-u复合材料,首次充电比容量为1 611.4 mAh/g,在之后的循环中充电比容量下降剧烈,200次循环后充电比容量仅有537.7 mAh/g,容量保有率仅为33.3%。以三聚氰胺作为前驱体的Si@C3N4-m复合材料首次充电比容量达到1 696.2 mAh/g,200个循环后充电比容量为802.3 mAh/g,容量保有率为47.3%。以硫脲作为前驱体的Si@C3N4-t复合材料首次充电比容量达到1706.4 mAh/g,200个循环后充电比容量为971.7 mAh/g,容量保有率为56.9%。可以看出,三聚氰胺和硫脲作为前驱体得到的复合材料循环性能都较好,Si@C3N4-t的循环保持率较高。这可能是由于Si@C3N4-u的片层结构松散且薄,在循环过程中,C3N4-u不足以限制Si,使其从集流体脱落,阻碍电子的传导。而在Si@C3N4-t复合材料中,Si部分夹嵌在C3N4-t的片层结构以及大孔洞中,这种厚大的片层结构以及孔洞不仅能够提高Si颗粒的分散性,增加其与电解液的接触面积,而且还能较好的缓解硅的体积膨胀,从而减少活性物质和电极的粉碎,因此表现出较稳定的循环性能。图10(b)为Si@C3N4复合材料在200 mA/g的电流密度下的首次充放电曲线。从图中可以看出,在首次放电过程中,放电曲线在1.3~0.2 V之间呈现一个缓和的斜坡,对应材料表面SEI膜的形成。在0.1 V左右有一个较长的放电平台,对应si的嵌锂过程[25]。Si@C3N4-u、Si@C3N4-m、Si@C3N4-t的首圈库伦效率分别为71.27%、80%、80.28%。 Si@C3N4复合材料的倍率性能如图10(c)所示,Si@C3N4-u复合材料在100 mA/g的电流密度下的首圈充电比容量为1 411.6 mAh/g,随着电流密度的提高,容量开始衰减。当电流密度增加到1 A/g,材料的比容量为726 mAh/g,当电流密度增加到2 A/g后,材料的比容量仅为472.8 mAh/g。当电流密度恢复到100 mA/g时,电池的比容量恢复到1 156.7 mAh/g,约占初始容量的81.9%。Si@C3N4-t表现出较Si@C3N4-u与Si@C3N4-m优越的倍率性能,其在100,200,500,1和2 A/g的电流密度下,平均可逆比容量分别为1 585.6,1 374.1,1 188.9,995.6和750.5 mAh/g,呈现出较好的充放电性能。当电流密度重新恢复到100 mA/g时,Si@C3N4-t复合电极的充电比容量可达1 407 mAh/g,表明Si@C3N4-t复合材料结构相对稳定。
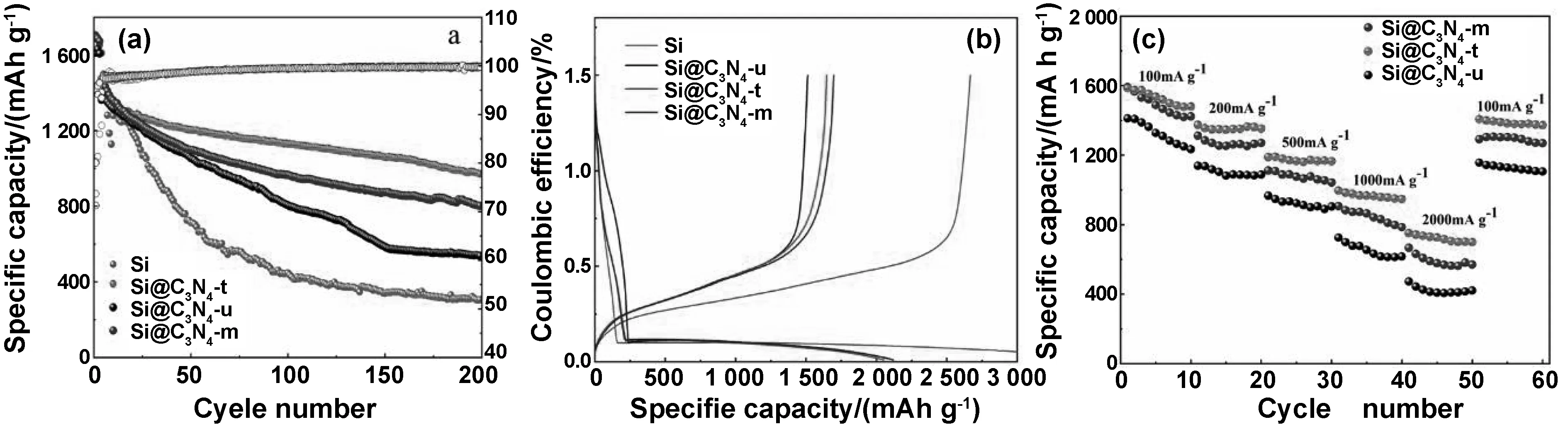
图10 复合材料的循环性能曲线(a)、首次充放电曲线(b)、倍率性能曲线(c)Fig 10 Cycling performances curve, initial charge and discharge curve and rate performance curve of compound material
3 结 论
采用尿素、三聚氰胺、硫脲三种前聚体通过热缩聚法成功制得C3N4,再用超声法与硅复合得到3种负极材料,结论如下:
(1)以硫脲为前聚体制得的Si@C3N4-t材料结晶度良好,硅颗粒可以进入片层以及大的孔洞中,有利于缓解材料充放电过程中造成的结构塌陷问题,提高材料的结构稳定性。
(2)Si@C3N4-t表现出优于其它两种材料的电化学性能,该复合电极在500 mA/g的电流密度下,200次充放电循环后可逆容量为 971.7 mAh/g,具有较好的比容量和电化学稳定性。
