Data Comparison and Software Design for Easy Selection and Application of CRISPR-based Genome Editing Systems in Plants
2021-08-04YiWangFatmaLecourieuxRuiZhangZhanwuDaiDavidLecourieuxShaohuaLiZhenchangLiang
Yi Wang ,Fatma Lecourieux ,Rui Zhang ,Zhanwu Dai ,David Lecourieux ,Shaohua Li ,Zhenchang Liang
1Beijing Key Laboratory of Grape Science and Enology,and CAS Key Laboratory of Plant Resources,Institute of Botany,the Innovative Academy of Seed Design,Chinese Academy of Sciences,Beijing 100093,China
2University of Chinese Academy of Sciences,Beijing 100049,China
3Ecophysiology and Functional Genomics of Vine,Bordeaux Sciences Agro,INRAE,University of Bordeaux,Institute of Vine and Wine Sciences Bordeaux-Aquitaine,Villenave d’Ornon 33140,France
4College of Plant Protection,Shandong Agricultural University,Taian 271018,China
5Sino-Africa Joint Research Center,Chinese Academy of Sciences,Wuhan 430074,China
Abstract CRISPR-based genome editing systems have been successfully and effectively used in many organisms.However,only a few studies have reported the comparison between CRISPR/Cas9 and CRISPR/Cpf1 systems in the wholegenome applications.Although many web-based toolkits are available,there is still a shortage of comprehensive,userfriendly,and plant-specific CRISPR databases and desktop software.In this study,we identified and analyzed the similarities and differences between CRISPR/Cas9 and CRISPR/Cpf1 systems by considering the abundance of proto-spacer adjacent motif(PAM)sites,the effects of GC content,optimal proto-spacer length,potential universality within the plant kingdom,PAM-rich region(PARR)inhibiting ratio,and the effects of G-quadruplex(G-Q)structures.Using this information,we built a comprehensive CRISPR database(including 138 plant genome data sources,www.grapeworld.cn/pc/index.html),which provides search tools for the identification of CRISPR editing sites in both CRISPR/Cas9 and CRISPR/Cpf1 systems.We also developed a desktop software on the basis of the Perl/Tk tool,which facilitates and improves the detection and analysis of CRISPR editing sites at the whole-genome level on Linux and/or Windows platform.Therefore,this study provides helpful data and software for easy selection and application of CRISPR-based genome editing systems in plants.
KEYWORDS CRISPR/Cas;PAM-rich region;G-Q;Software;Database
Introduction
Clustered regularly interspaced short palindromic repeat(CRISPR)/CRISPR-associated proteins (Cas) system is derived from the adaptive immune system of prokaryotes,and has been adapted as an effective tool for plant genome editing[1−3].It is classified into two classes:class 1 uses a complex of multiple Cas proteins to degrade foreign nucleic acids,and class 2 uses a single large Cas protein for the same purpose.Several CRISPR/Cas systems have been developed using class 2 system,and amongst the most popular ones are Cas9 (especially fromStreptococcus pyrogene) and Cpf1 (fromPrevotellaandFrancisella1)systems.Since the first report on CRISPR/Cas9-directed genome editing inArabidopsisand tobacco,its use has been widely applied in many plant species,such as rice,wheat,maize,tomato,potato,cotton,soybean,grape,apple,and poplar[3−8].CRISPR/Cpf1 has been recently reported as a new CRISPR-based genome editing system and has also been successfully applied in many plant species [9−11].
Both CRISPR/Cas9 and CRISPR/Cpf1 are guided by guide RNAs (gRNAs) which enable them to edit genomes precisely and accurately.However,though the two systems have a common origin,there are some notable differences.For example,the CRISPR/Cas9 system recognizes GC-rich proto-spacer adjacent motif (PAM) sequences and cuts double-stranded DNA,thereby generating blunt-end double-stranded breaks (DSBs).It is mainly used for gene knockout by inducing small insertions and deletions(indels)[12,13].In contrast,the CRISPR/Cpf1 system is simpler because it recognizes T-rich PAM sequences and produces staggered cuts,thereby leaving 5-nt 5′ overhangs.These overhangs can be used to generate larger indels than those produced by the CRISPR/Cas9 system [2].Cpf1 is also a ribonuclease that only requires one CRISPR RNA(crRNA)without atrans-acting crRNA(tracrRNA)[1].
Previous studies have reported that both PAM-rich traffic light reporter (TLR) sites and G-quadruplex (G-Q) structures present in the PAM regions interfere with the CRISPR/Cas9 editing efficiency[14,15].The presence of more than three PAMs on the target strand,more than four PAMs on the opposite strand,and a combination of two PAMs on the target strand and three PAMs on the opposite strand inhibits Cas9 editing[15].G-Q structures that refer to GC-rich DNA sequences can also limit the transfection efficiency[15,16].
With the development of genome editing systems,a series of CRISPR-related databases and web-based tools,including Cas-Database [17],WGE [18],CrisprGE [19],CRISPR-P 2.0 [20],Crispr-GE [21],and Grape-Crispr [22],have been constructed.However,to date,only a few tools can be used to design gRNAs and test their effectiveness by using platforms,such as Windows and Linux,in plants.In this study,we aim to design an effective automatic desktop software interface that allows users to identify and select specific proto-spacers on the basis of the whole-genome sequences in plants.
Results
Identification of PAM sites for CRISPR/Cas9 and CRISPR/Cpf1 systems
PAMs for both CRISPR/Cas9 and CRISPR/Cpf1 systems were identified and analyzed in 138 plant genomes.A total of 11,441,028,628 and 25,066,950,663 putative PAMs were identified for CRISPR/Cas9 and CRISPR/Cpf1 respectively throughout the genomes (Table S1),with an average of 82,376 putative PAMs per mega base (Mb) for CRISPR/Cas9 and 175,201 putative PAMs per Mb for CRISPR/Cpf1.Notably,the number of CRISPR/Cpf1 potential PAMs was two folds that of CRISPR/Cas9.Among all proto-spacers of these PAMs,the rates of potential specific proto-spacers(with no mismatch or gap when applied against plant genomes) were 63.64% for CRISPR/Cas9 and 74.45% for CRISPR/Cpf1.According to this analysis,the numbers of CRISPR/Cas9 and CRISPR/Cpf1 editing sites were linearly correlated with genome size (Figure 1A).The correlation coefficients (R2) were higher than 0.98 for all potential editing sites.For potential specific editing sites for CRISPR/Cas9 and CRISPR/Cpf1,theR2coefficients were 0.874 and 0.952,respectively.
Next,the relationship between genome PAM composition and GC content was analyzed.For both CRISPR/Cas9 and CRIPSR/Cpf1 systems,the ratios of each PAM type(Figure 1B) were almost equal on both DNA strands,and the ratio of PAM sites on one strand was almost identical to that on the other strand within the same genome.In addition,the percentage of PAM types was highly affected by the GC content.In the CRISPR/Cas9 system,a positive correlation between the GC content and four PAM types(CGG,GGG,CCG,and CCC) was observed (Figure 1B).This result was particularly true for CGG and CCG sites.The same analysis was carried out for the CRISPR/Cpf1 system,and we found that the GC-rich PAMs(CAA,GAA,TTC,and TTG) were also positively correlated with the genome GC content (Figure 1B).
In the CRISPR/Cas9 system,the high GC content was translated to a high potential to edit the target sites and a high density as well.By contrast,the potential and specific editing sites of the CRISPR/Cpf1 system were highly negatively correlated with the GC content.Although GC content considerably affected the abundance of PAM sites,in most situations,the number of CRISPR/Cpf1 PAMs was generally higher than that of CRISPR/Cas9 PAMs.Plants contained a relatively narrow range of GC content (30%–50%) (Figure 1C),suggesting that the G-and C-biased PAMs were considerably less than the A-and T-biased PAMs.When the GC rate narrowed to 50%,the abundances of the CRISPR/Cas9 and CRISPR/Cpf1 PAM sites were similar.

Figure 1 Identification and analysis of CRISPR/Cas9 and CRISPR/Cpf1 PAM sites in plant genomes
Determination of optimal proto-spacer length
The length of proto-spacers is an important factor in the CRISPR/Cas system.In this study,considering some smallsized genomes,we selected a proto-spacer length ranging from 13 nt to 40 nt.“13 nt”was selected because if the base pairs were distributed randomly,the 13-nt spacer was specific in the 67-Mb genomes as 413=67,108,864.The results showed that a proto-spacer of 20 nt(the one generally used)was specific enough for both CRISPR/Cas systems(Figure2).Most of the studied species reached their threshold value at the length between 16 nt and 18 nt.Increasing the protospacer length only offered a minute advantage on the ratio of specific proto-spacers to all potential proto-spacers.However,Solanum lycopersicumshowed a different trend.Although the threshold value of this species was 20 nt,it was strongly enhanced by increasing the proto-spacer length.In addition,species with small genomes,such asArabidopsis thalianaandVolvox carteri,possessed a relative higher ratio of specific proto-spacers than species with large genomes.
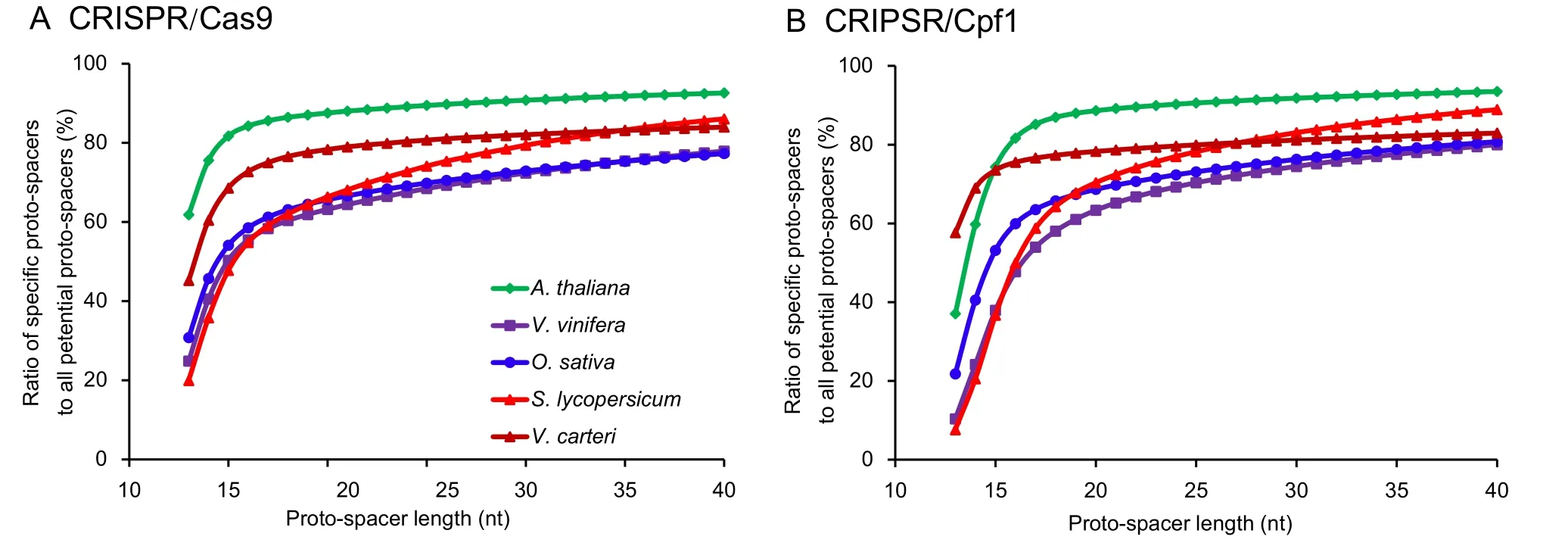
Figure 2 Effect of proto-spacer length on the ratio of specific proto-spacers
Potential universality of proto-spacer sequences within the plant kingdom
Identification of universal proto-spacer sequences can help to edit genes in multiple plant species in an efficient and convenient way.According to our homology analysis,the similarity of the proto-spacer sequences was linked to the relationship between the plants studied.For CRISPR/Cas9 system,without considering editing limitations (e.g.,gene structure limitations),the proto-spacer sequences of a certain species can only be used for its closest relatives,and homologous proto-spacer sequences only existed in small and limited areas (Figure 3A−D).Analysis of specific CRISPR/Cas9 proto-spacers in all 138 tested plants showed that these specific proto-spacers displayed low homology(Figure 3E).In monocots,two CRISPR/Cas9 proto-spacer clusters showed high homology(Figure 3B):one containedOryzaand its relatives,and the other containedTriticumand its relatives.InOryza,an average of 44.54% (45.02% and 44.06%) of the CRISPR/Cas9 proto-spacer sequences can be used equally between two rice modelsO.sativaL.ssp.japonicaandO.sativaL.var.9311.On average,66.74%(41.27%–97.84%) of the CRISPR/Cas9 proto-spacer sequences were effective among theOryzaspecies.When considering the specific proto-spacer sequences identified inOryza,the average homology decreased to 61.81%(Figure 3F).InTriticum,the highest average homology of effective CRISPR/Cas9 proto-spacers between two species was 84.32% (82.15% and 86.49%),which was identified between the two accessions ofT.turgidumL.ssp.durum.On average,46.31% (25.01%–86.49%) of the CRISPR/Cas9 proto-spacers can be used among theTriticumspecies.Considering the specific CRISPR/Cas9 proto-spacer sequences inTriticum,the average homology dramatically decreased to 12.69% (Figure 3F).In Brassicaceae and Solanaceae families,only 6.71% and 4.17% of the CRISPR/Cas9 proto-spacers can be effective among their species/varieties,respectively(Figure 3C and D).Among the seven sequencedNicotianagenomes,only 32.24%and 37.98%of the CRISPR/Cas9 proto-spacer sequences can be used in the whole Solanaceae family and in theNicotianagenus,respectively.However,the CRISPR/Cas9 proto-spacer sequences identified from threeN.tabacumL.varieties can be efficiently used to edit each other with an average homology of 97.80%.The CRISPR/Cas9 proto-spacers identified from three wild tobacco varieties that were considered to be parents ofN.tabacumwere also effective in editing three modern tobacco cultivars (N.otophora:64.88%;N.tomentosiformis:87.20%;andN.sylvestris:92.89%).In addition,only 19.80% of the CRISPR/Cas9 proto-spacers from the model plantN.benthamianacan be used to edit otherNicotianavarieties.Considering the specific protospacers,only 7.21% and 14.12% of the specific CRISPR/Cas9 proto-spacers respectively identified from Brassicaceae and Solanaceae can target species belonging to their own family(Figure 3G and H).
The proto-spacer homology analysis was also performed for the CRISPR/Cpf1 system,we found similar results to those observed for the CRISPR/Cas9 system(Figure 3I–P).High homology also existed in some closely related species.A similar trend was also found inOryza,Triticum,Brassicaceae,and Solanaceae.InOryzaandTriticum,the average homologies of 32-nt proto-spacers were 57.15% and 26.59%,respectively,and the average homologies of specific proto-spacers were 53.83% and 12.36%,respectively.In Brassicaceae,the average homologies of proto-spacers and specific proto-spacers were only 4.31% and 2.37%,respectively.In Solanaceae,the average homologies of proto-spacers and specific proto-spacers were 14.88% and 11.89%,respectively.InNicotiana,on average,42.05% of the proto-spacers and 28.57% of the specific proto-spacers were homologous.
PAM-rich regions in CRISPR/Cas9 and CRISPR/Cpf1 systems
Next,the PAM-rich regions (PARRs) that might inhibit CRISPR/Cas9-mediated genome editing were analyzed in five species,includingA.thaliana,Vitis vinifera,S.lycopersicum,O.sativa,andV.carteri.As shown inFigure 4A,the inhibiting ratio ranged from 2.97%to 23.63%(9.75%on average),with highest inhibiting ratios observed in monocotO.sativaand algaeV.carteri.Further analysis of eight dicots and eight monocots revealed a significant higher PARR inhibiting ratio in monocots than in dicots (Figure 4B;Table S2).
Although the effect of the PARR inhibition in the CRISPR/Cpf1 system has not been reported to date,we considered it based on the similarity present in both systems.In contrast to the CRISPR/Cas9 system,the PARR inhibiting ratios observed for the CRISPR/Cpf1 system were considerably higher in dicot plants (A.thaliana,V.vinifera,andS.lycopersicum) than those in monocotO.sativaand algaeV.carteri(Figure 4 C;Table S2).Further analysis of eight monocots and eight dicots also revealed a significant higher PARR inhibiting ratio in dicots than in monocots(Figure 4D;Table S2).
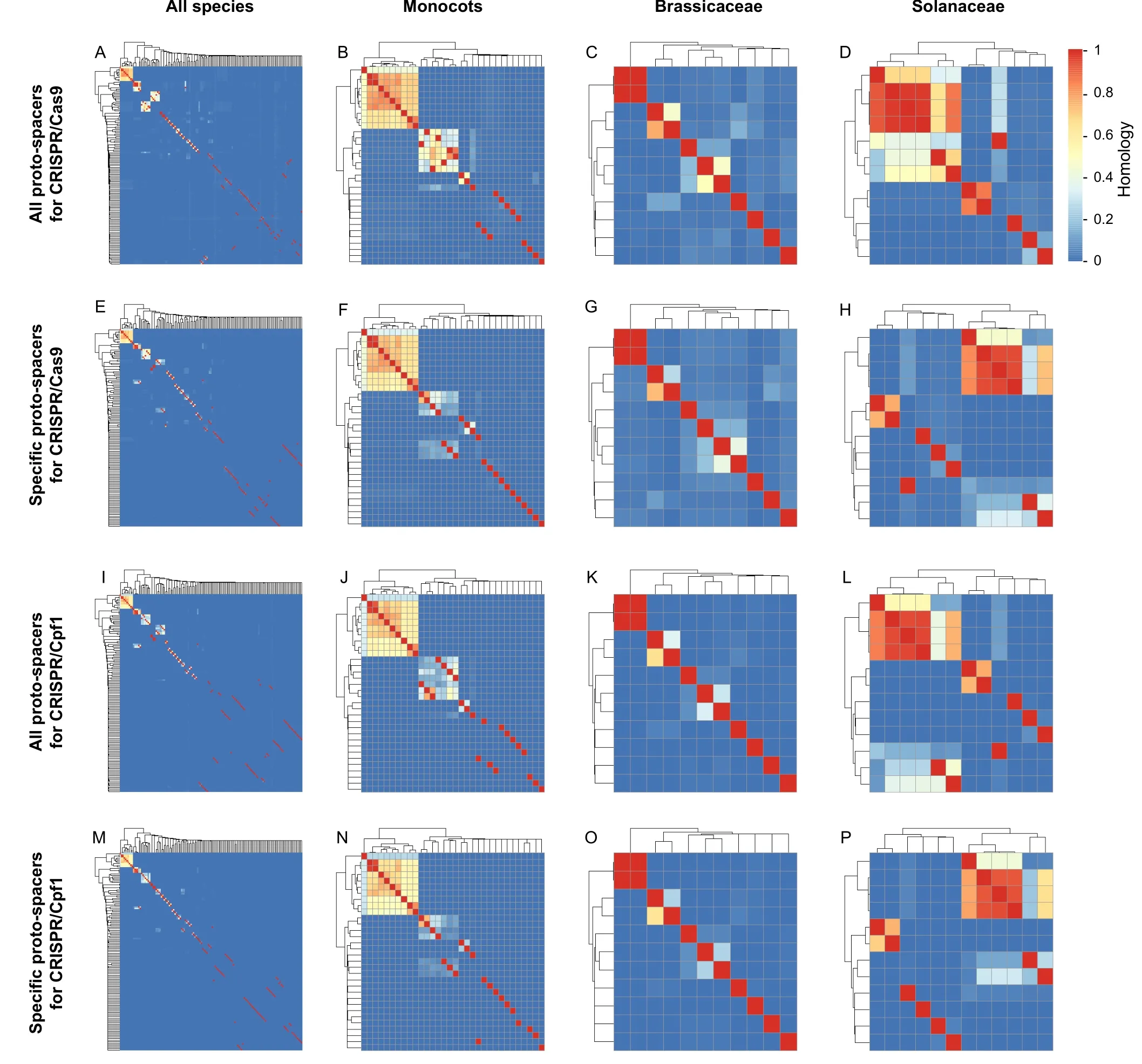
Figure 3 Homology analysis of proto-spacer sequences for CRISPR/Cas systems

Figure 4 Analysis of PARR inhibiting ratio for CRISPR-mediated editing in plants
Influence of G-Q structures on the effectiveness of CRISPR-mediated editing
According to the G-Q analysis,on average,32.87% of CRISPR/Cas9-driven editing sequences were located in the potential G-Q structure regions,although most of these sequences were located in two G-tetrads (Table 1).Disparities existed amongst different species studied here,with monocotO.sativaL.spp.japonicaand algaeV.carterishowing highest G-Q influence rates(42.63%and 63.10%,respectively).
In CRISPR/Cpf1 system,the average G-Q influence rate was 15.23%,which was less than half of the estimated value for CRISPR/Cas9-driven editing sites.The highest influence rate was obtained inV.carteri(40.47%),and the lowest was observed inS.lycopersicum(6.38%;Table 1).

Table 1 G-Q composition and influence rate in CRISPR/Cas9 and CRISPR/Cpf1 systems
Identification of genes that can be edited by CRISPR/Cas systems
In this study,the proto-spacers that were located in PARRs or in G-Q regions were considered as ‘useless’proto-spacers,while other proto-spacers were considered as high-quality proto-spacers.We proposed that genes lacking high-quality proto-spacers could not be edited.Eventually,we identified 27,678 genes inA.thaliana,25,325 genes inV.vinifera,34,451 genes inS.lycopersicum,54,276 genes inO.sativa,and 14,059 genes inV.carteri,which could be edited by the CRISPR/Cas9 system(Figure 5,Figure S1A–E).Moreover,Venn analysis of the Gene Ontology (GO)terms of genes that could not be edited by the CRISPR/Cas9 system showed an insignificant correlation between gene function and plant genome ability to be processed by CRISPR/Cas9 system(Figure S1F).
Given that no report exists on the barriers to the CRISPR/Cpf1 genome-editing system,in this study,we only considered the presence of G-Q structures and the specificity of proto-spacers.The CRISPR/Cpf1 system was tested for its editing efficiency with respect to CRISPR/Cas9 system.As shown in Figure 5,most genes could be edited by both systems,with some specifically edited by either CRISPR/Cpf1 or CRISPR/Cas9 system(Figure 5).As an example,inA.thaliana,27,643 genes could be edited by both CRISPR/Cpf1 and CRISPR/Cas9 systems,with 346 genes specifically edited by CRISPR/Cpf1 system and 35 genes specifically edited by CRISPR/Cas9 system.Notably,142−1194 genes could not be edited by any of these two CRISPR/Cas systems in the five analyzed species.

Figure 5 Comparison of CRISPR/Cas9 and CRISPR/Cpf1 systems in five plant genomes
PLANT-CRISPR database
On the basis of this large amount of data,a database named PLANT-CRISPR was established.This database contains three main parts.The first part corresponds to the CRISPR search.Here people can search any potential CRISPR/Cas editing sites in the genomes of 138 plants.The result of this search provides the GC content,the predicted PARRs and G-Q structures,and related gene information linked to these CRISPR/Cas editing sites.In the second part,we developed two web tools.The first tool can design gRNAs for the uploaded sequence.This tool contains some parameters such as the PAM type,proto-spacer length,and GC content.The second tool provides a web-based effective computational method to test the effectiveness of the designed gRNAs.This tool should set the acceptable mismatches and the optimal proto-spacer length.The third part provides download links to all data generated in this study,and all these data can be used freely.
CRISPR/Cas detection and its effective software
Unlike other database and web-based tools,we also provided a desktop software application.This software is based on Perl language,and the interface is provided by the Perl-Tk module.The first part (CRISPR_detector) detects potential CRISPR/Cas editing sites and annotates them with the annotation files (GFF3).This software deals with the genome size data in a relatively short time and uses a short computational source (Table S3).The second part(Elec-CRISPR) provides a local effective test for protospacers on the basis of the reference genome;when necessary,high-throughput sequencing data can also be used as a reference.The speed of this software is also influenced by the genome size(Table S3).Single chromosome or scaffold longer than 1 Gb was unsupported by this software.The software can be downloaded at www.grapeworld.cn/pc/index.html.The efficiencies of the two scripts were tested on Linux and Windows platforms(Table S3),respectively.
Discussion
High density of PAM sites and influence of GC content
In this study,on average 82,376 and 175,201 putative PAM sites per Mb are identified for CRISPR/Cas9 and CRISPR/Cpf1 systems,respectively.Hence,in a sequence,a CRISPR/Cas9 editing site and a CRISPR/Cpf1 editing site can be found every 12.14 bp and 5.71 bp,respectively.Therefore,both CRISPR/Cas systems can edit the genome accurately and almost everywhere.
In plant genomes analyzed,the abundance of CRISPR/Cpf1 editing sites is considerably higher than that of CRISPR/Cas9.CRISPR/Cpf1 can also target more areas than CRISPR/Cas9 mainly due to the fact that the GC content in most plant genomes ranges between 30% and 50% (less than half of the genome).By contrast,AT base pairs are abundant,leading to a high rate of NAA/TTN motifs that correspond to the CRISPR/Cpf1 PAM sites.In the present study,GC content is identified as an important factor which influences the abundance of CRISPR/Cas editing sites and the PAM composition.High GC content can increase GC-rich PAM abundance.Considering that the GC content in most plant genomes observed is lower than 50%,CRISPR/Cpf1 editing tends to occur in plants.Therefore,CRISPR/Cpf1 editing system appears to be a new effective method for genome engineering.In addition,the combination of both CRISPR/Cas methods can improve plant genome editing.These results also need to consider the fact that the CRISPR/Cas systems evolve and offer additional Cas9 and Cpf1 orthologs that can improve genome editing.Thus,although Zetsche et al.[1] has reported that FnCpf1 recognizing a TTN PAM is inefficient for human cell editing,other studies have identified new efficient Cas proteins.For example,Kim et al.[9] showed that the Cpf1 orthologs AsCpf1 and LbCpf1 targeting either TTTN or TTTV PAM sequences (instead of TTN) can be applied to edit a mammalian cell genome.Hu et al.[23]developed an expanded PAM SpCas9 variant(xCas9),which not only can recognize a broad range of PAM sequences,including NG,GAA,and GAT,but also has much greater DNA specificity than SpCas9.All these results suggest that the evolved systems should increase PAM compatibility and DNA specificity,thus leading to efficient and accurate genome editing.
Photo-spacers of 20 nt in length are not the best for both CRISPR/Cas editing systems
A 14-nt read typically loses its randomness in a 200-Mb genome (414=268,435,456),and in a 20-Gb genome,this number should be 18 nt.However,considering genome-wide duplication and other genome events,it must be more complex than the random conditions.In this study,to provide a guidance to the optimal proto-spacer length for CRISPR/Cas9 and CRISPR/Cpf1 systems,we calculated the ratios of specific proto-spacers to all potential proto-spacers at different proto-spacer lengths.Both CRISPR/Cas systems reach their threshold values at 16–18 nt.Proto-spacer length longer than 18 nt only shows a slow increase of the ratio.Thus,the currently used 20-nt spacer is long enough to maintain the specificity within the genome.In some species,such as tomato,a suitable extension of the proto-spacer length can still improve the ratio.Therefore,when necessary,an adjustment of the proto-spacer length is also needed.However,one should keep in mind that the optimal proto-spacer length depends on several factors,including experimental conditions,biological material,and Cas protein types.
Homologous proto-spacers can be used in limited areas
The homology analysis of the proto-spacer sequences among 138 plant genomes indicates that the proto-spacer sequences selected for genome editing in one species can also be used in some close relatives,such as inOryzaspecies and in tobacco cultivars.The high homology betweenNicotianaspecies also suggests a relationship between cultivars and three wild-type plants.The low homology betweenN.benthamianaand otherNicotianavarieties implies thatN.benthamianamay have a specific position inNicotianagenus.These results suggest that most proto-spacers selected for genome editing can be used directly within the same genus,with the exception of few particular species.Thus,the proper use of proto-spacer sequences within species could reduce time and avoid some unnecessary shortcomings.
PARR is an important limitation for CRISPR/Cas system
The analysis of PARRs in CRISPR/Cas9 system indicates that PARR density influences~10%of the potential editing sites.The PARR inhibiting ratios in monocotO.sativaand algaeV.carteri.are higher than those of dicotA.thaliana,Vitis vinifera,S.lycopersicum.Comparison of eight dicots and eight monocots also indicates a significantly higher PARR inhibiting ratio in monocots than in dicots.Therefore,when editing monocot and algae genomes using CRISPR/Cas9 system,PARRs should be considered carefully.
The influence of PARRs in CRISPR/Cpf1 system has not been reported yet.Considering that CRISPR/Cpf1 also belongs to the CRISPR/Cas system and its editing principles are similar to those of CRISPR/Cas9,we proposed that PARRs should also influence the editing efficiency of CRISPR/Cpf1.In contrast to CRISPR/Cas9 system,a significantly higher PARR inhibiting ratio was observed in dicot plants,suggesting that dicot plants may bear more disturbance from PARRs in CRISPR/Cpf1 system than in CRISPR/Cas9 system.
For CRISPR/Cas9 and CRISPR/Cpf1 systems,the significant difference in PARR inhibiting ratios between monocots and dicots suggests that the unbalanced distribution of PARRs in monocots and dicots may be influenced by the genome characteristics.Similarly,a previous study comparing simple sequence repeats in monocots and dicots has revealed huge differences in the GC/AT ratio[24].
A previous study using PAM-rich TLRin vivohas shown the inhibition effect of the PARRs on CRISPR/Cas9 editing efficiency[15].In the present study,the large-scale analysis shows that PARR is an important limitation for CRISPR/Cas genome editing.This influence also varies with the plant features.
G-Q is common among the editing sites
G-Q structure influences 32.87% of the CRISPR/Cas9 editing sites and 15.23% of CRISPR/Cpf1 editing sites,indicating that G-Q may highly inhibit the editing efficiency due to their special structures.For both CRISPR/Cas systems,the influence of G-Q structures in dicots is not as important as that observed in monocots and algae.In CRISPR/Cas9 system,more than 40% editing sites ofO.sativaL.spp.JaponicaandV.carteriare potentially influenced by G-Q structures,which are considerably higher than those of dicots.This trend was also observed in the CRISPR/Cpf1 system.Thus,like PARRs,G-Q structures are also influenced by the genome characteristics of species.High GC ratio may lead to increased G-Q structures in monocot genomes.Some studies have also suggested that the chromatin structure near the target sites plays an important role and affects the accessibility and efficiency of the CRISPR/Cas system [25].However,given that the chromatin structure is dynamic and difficult to obtain,we only focus on PARRs and G-Q structures.
CRISPR/Cas system can edit most plant genes
Considering several limitations,both CRISPR/Cas9 and CRISPR/Cpf1 systems can edit most plant genes.However,several hundreds of genes cannot be edited by CRISPR/Cas9 system,indicating that this effective editing tool is not totally efficient.The new CRISPR/Cpf1 system is also identified as an extremely effective tool for plant genome editing,and to some extent,it can edit some genes that are not targeted by the CRISPR/Cas9 system.However,similar to CRISPR/Cas9 system,it does not target all plant genes.Hence,combining these two editing tools may improve the plant genome editing efficiency by targeting increased number of genes.In addition,as several hundreds of genes are still refractory to genome editing,improving the existing tools or producing new editing tools that can target most or all plant genes is necessary.Currently,many studies are on the process of developing new or improved existing Cas proteins that can target various PAM sequences.
Highly effective database and software
PLANT-CRISPR (www.grapeworld.cn/pc/index.html) is the largest CRISPR/Cas-related database to date,which contains 138 plant genomes and provides accurate and detailed information for CRISPR/Cas9-and CRISPR/Cpf1-mediated genome editing.The database can 1)supply all the proto-spacers contained in the genomes,2)provide specific statistics,GC content,detailed annotation,and some other information,and 3)offer additional choices for researchers,making plant genome editing more accurate and flexible.At the same time,the possibility of designing gRNAs and primers for potential editing sites can make genome editing easier for related species.The database also provided two web tools:one is for designing gRNAs for the uploaded sequence,and the other aims to test the effectiveness of the designed gRNAs.Currently,138 plant genomes can be processed by these tools;however,if users supply new genome sequences,such sequences will be added to the database.
The desktop software,consisting of CRISPR_detector and Elec-CRISPR,can process a considerable number of genome sequences within a short period of time,following users’ expectations.This software is more efficient and convenient than previous/older tools and software (Table S3).Numerous sequences or proto-spacers can be handled simultaneously,and people can choose their own favorite sequences or reference genomes.The parameters of the software provide choices in designing gRNAs and performing efficiency test.This software will be updated to some more user-friendly languages in the near future.
Although many efficient database and web tools have been published,our work provides several new features.First,our database contains information regarding 138 plant genomes.Second,this database provides an interface software on the basis of the Perl-Tk,thereby making related analyses more convenient and effective.Last,our work allows a more flexible analysis to predict the CRISPR/Cas editing sites,as some parameters can be modified/adapted by each user.
PLANT-CRISPR is part of the grapeworld platform(http://www.grapeworld.cn/) and will be updated annually.All new published genomes will be considered.Users also can suggest and supply the genome or other data which can be used in this database to us,and all these suggestions will be considered carefully.
Conclusion
In this study,we identify the CRISPR/Cas9 and CRISPR/Cpf1 editing sites in 138 plant genome sequences.The comparative analysis of proto-spacer sequences shows that GC content is an important factor influencing the abundance of CRISPR/Cas9 and CRISPR/Cpf1 editing sites.In plants,a 20-nt proto-spacer is sufficient for most genomes,and a slight adjustment should be acceptable for some species.Homology analysis shows that proto-spacers that are selected for gene editing for some species can be directly used in other plants from the same genus.The PARRs and G-Q structures are common among the proto-spacers and should be considered seriously.The existing CRISPR/Cas systems can edit most plant genes but not all.Therefore,developing and improving tools are necessary to make plant genome editing more accurate and flexible.Finally,in the frame of the ongoing development,the PLANT-CRISPR database and software that we developed can considerably contribute to improving and facilitating plant genome editing studies.
Materials and methods
Genome sequences used in this study
All genome sequences used in this study were downloaded from public databases.Detailed information is listed in Table S1.
Analyses of PAMs,proto-spacers,and GC content
The identification and analyses of PAMs,proto-spacers,and GC content were processed by the Perl scripts.According to the related studies,the PAM sequences analyzed in this study were NGG for CRISPR/Cas9 and NAA for CRISPR/Cpf1.In each system,the PAM sequences were classified into four types (i.e.,AGG,TGG,CGG,and GGG for CRISPR/Cas9;AAA,TAA,CAA,and GAA for CRISPR/Cpf1) to explore the composition and correlation with the GC content.The GC content was calculated by Perl scripts on the basis of the genome sequences.Homology of protospacers among different species was analyzed with 10,000 proto-spacers from each species,and the proto-spacers from a certain species were searched against all other species.Neither mismatch nor gap was allowed among the editing sites.
Identification of PARRs and G-Q structures
The PARRs for the CRISPR/Cas9 system were identified similarly to the study of Malina and colleagues [15].In brief,more than 3 PAMs on the target strand,more than four PAMs on the opposite strand,and a combination of two PAMs on the target strand and three PAMs on the opposite strand were identified as inhibitory PARRs.In the CRISPR/Cpf1 system,PARRs were identified by their PAMs(NAA or TTN).For the 32-nt proto-spacers selected in this study,more than five PAMs on the target strand and more than six PAMS on the opposite strand were identified as inhibitory PARRs for CRISPR/Cpf1 system.
According to previous studies [26−28],the G-Q model used in this study is Gx-Ny-Gx-Nz-Gx-Nr-Gx(where N=A,T,C,or G;x=2–4;y=1–10;z=1–10;r=1–10).The G-Q structure was identified using a Perl script.All editing sites were annotated with the detailed G-Q information or non-G-Q.
Venn analysis
The Venn analysis was processed using“jvenn”(http://jvenn.toulouse.inra.fr/app/index.html) [29],and the GO analysis was performed using the AgriGo database (http://systemsbiology.cau.edu.cn/agriGOv2/index.php)[30].
Database architecture and web interface
The PLANT-CRISPR database (www.grapeworld.cn/pc/index.html)was constructed by HTML,PHP,and MYSQL,and some functions were realized using Perl scripts.The interface was written using HTML and CSS.The user inquiries were uploaded to the system and processed by PHP and MYSQL or Perl scripts.All the obtained data,including the software used in this study,were stored in this database.
For the web tools and desktop software interface,the offtarget effect was identified by the mismatches on the target sequences.The target sequences were classified into two types,namely,the high-fidelity and the rest sequences.No mismatch or gap was allowed on the high-fidelity sequences,while mismatches or gaps were allowed on the rest sequences according to the user selection.Users with only whole-genome sequencing data can convert FASTQ format into FASTA format by using the supplied Perl scripts and then verify the proto-spacer sequences on the basis of the Elec-CRISPR module.
Code availability
The datasets and software generated and/or analyzed in this study are available in the PLANT-CRISPR (http://www.grapeworld.cn/pc/index.html).
CRediT author statement
Yi Wang:Formal analysis,Writing-original draft.Fatma Lecourieux:Methodology,Writing-review&editing.Rui Zhang:Formal analysis.Zhanwu Dai:Formal analysis.David Lecourieux:Writing -review &editing.Shaohua Li:Conceptualization,Methodology,Supervision.Zhenchang Liang:Conceptualization,Methodology,Supervision.All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
We thank Dr.Chong Ren for his critical suggestion in data analysis.This work was supported by grants from the National Key R&D Program of China (Grant No.2018YFD1000105),the National Science Foundation of China (Grant No.31772266),the Agricultural Breeding Project of Ningxia Hui Autonomous Region,China (Grant No.NXNYYZ20150203),and the Hundred Talents of the Chinese Academy of Sciences..
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.05.008.
ORCID
0000-0002-4534-8946 (Yi Wang)
0000-0003-1601-2500 (Fatma Lecourieux)
0000-0002-0897-7161 (Rui Zhang)
0000-0002-7625-8337 (Zhanwu Dai)
0000-0001-8703-8674 (David Lecourieux)
0000-0001-7707-206X (Shaohua Li)
0000-0002-4644-4454 (Zhenchang Liang)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- TSUNAMI:Translational Bioinformatics Tool Suite for Network Analysis and Mining
- Identifying Novel Drug Targets by iDTPnd:A Case Study of Kinase Inhibitors
- QAUST:Protein Function Prediction Using Structure Similarity,Protein Interaction,and Functional Motifs
- Computational Assessment of Protein–protein Binding Affinity by Reversely Engineering the Energetics in Protein Complexes
- eTumorMetastasis:A Network-based Algorithm Predicts Clinical Outcomes Using Whole-exome Sequencing Data of Cancer Patients
- BrcaSeg:A Deep Learning Approach for Tissue Quantification and Genomic Correlations of Histopathological Images