QAUST:Protein Function Prediction Using Structure Similarity,Protein Interaction,and Functional Motifs
2021-08-04FatimaZohraSmailiShuyeTianAmbrishRoyMeshariAlazmiStefanAroldSrayantaMukherjeeScottHeftyWeiChenXinGao
Fatima Zohra Smaili ,Shuye Tian ,Ambrish Roy ,Meshari Alazmi ,Stefan T.Arold ,Srayanta Mukherjee ,P.Scott Hefty,Wei Chen,Xin Gao
1Computational Bioscience Research Center (CBRC),Computer,Electrical and Mathematical Sciences and Engineering (CEMSE)Division,King Abdullah University of Science and Technology (KAUST),Thuwal 23955,Saudi Arabia
2Department of Biology,Southern University of Science and Technology of China (SUSTC),Shenzhen 518055,China
3Department of Computational Medicine and Bioinformatics,University of Michigan,Ann Arbor,MI 48109,USA
4College of Computer Science and Engineering,University of Ha’il,Ha'il 55476,Saudi Arabia
5Biological and Environmental Sciences and Engineering (BESE) Division,King Abdullah University of Science and Technology(KAUST),Thuwal 23955,Saudi Arabia
6Department of Molecular Bioscience,University of Kansas,Lawrence,KS 66047,USA
Abstract The number of available protein sequences in public databases is increasing exponentially.However,a significant percentage of these sequences lack functional annotation,which is essential for the understanding of how biological systems operate.Here,we propose a novel method,Quantitative Annotation of Unknown STructure(QAUST),to infer protein functions,specifically Gene Ontology (GO) terms and Enzyme Commission (EC) numbers.QAUST uses three sources of information:structure information encoded by global and local structure similarity search,biological network information inferred by protein–protein interaction data,and sequence information extracted from functionally discriminative sequence motifs.These three pieces of information are combined by consensus averaging to make the final prediction.Our approach has been tested on 500 protein targets from the Critical Assessment of Functional Annotation(CAFA)benchmark set.The results show that our method provides accurate functional annotation and outperforms other prediction methods based on sequence similarity search or threading.We further demonstrate that a previously unknown function of human tripartite motif-containing 22(TRIM22)protein predicted by QAUST can be experimentally validated.
KEYWORDS Protein function prediction;GO term;EC number;Protein structure similarity;Functionally discriminative motif
Introduction
As of today,over 150 million protein sequences are available in the UniProtKB/TrEMBL database [1].However,this increase in the number of known protein sequences does not reflect a parallel increase in our biological knowledge,as less than 1% of these sequences have a manually annotated function [2].On the other hand,the functional annotation of these sequences is not only an essential step for the understanding of physiological processes and biological systems in living entities,but also one of the highly challenging tasks in biology,which is why there is an increasing need to provide reliable,automated protein function annotation.
Significant efforts have been made to identify evolutionarily related proteins and automatically transfer functional annotations between homologous protein pairs [3–6].To make such sequence similarity-based functional transfer possible,powerful sequence-alignment methodologies have been developed.In particular,algorithms like BLAST/PSI-BLAST [3] and hidden Markov model (HMM)-based techniques [4–6] have been frequently used to transfer functional annotations between homologous proteins.The underlying assumption of these sequence-based methods is that evolutionarily related proteins may inherit the function of a shared common ancestor.However,there are numerous cases in which proteins with high sequence similarity have distinct functions [7,8].To partially address the problem,several methods have been developed to predict function using annotated conserved sequence motifs that are responsible for the functional aspect of the protein.These methods typically construct the sequence motifs from multiple sequence alignment of proteins belonging to the same protein family with known function [9–11].They,however,have two major limitations.First,high-quality sequence alignment is typically required for motif construction,which is not trivial to obtain especially when the sequence homology is low.Second,the accuracy is limited by the quality of functional annotation of motifs.To overcome these limitations,we propose in this work to use a protein-specific“functionally discriminative motif”constructed from sequence fragments excised from the template sequence.
From another perspective,the 3D structure of a protein sequence is believed to be more involved in its biological function [12,13],since structures are more conserved than the sequences.The 3D structure of a protein can therefore provide additional information for function transfer,especially when the sequence similarity between related proteins is too low for sequence homolog detection [14,15].However,the relationship between the protein function and its structure is not straightforward,as in some cases,similar structures perform the same function while in many cases similar folds perform different functions[16,17].Therefore,many prediction methods have been relying on local structure similarity search methods rather than global similarity search to identify functionally homologous proteins [18–20].Most of these approaches scan the query protein against a library of known conserved spatial motifs or known active sites (e.g.,binding sites) with known function [21].Local similarity search methods have been proven to be quite accurate in detecting functional similarity between proteins of different folds,but they also have a high probability of producing false positive matches [22].One possible solution is to combine global and local structure alignments to overcome the promiscuity of global structure comparison and low specificity of local structure matching[23,24],which we implement in this project.
A number of function prediction methods are based on the information extracted from protein–protein interaction(PPI)networks[25,26].The assumption in this case is that proteins that physically interact with each other frequently appear at the same sub-cellular location and are part of the same biological process[27].However,it is not always the case that proteins which interact with each other share the same molecular function [e.g.,Programmed cell death protein 1(PD1)and Programmed death-ligand 1(PD-L1)],which is why PPI information is not always sufficient to predict very specific functions[28].
Finally,recently there is an emergence of methods which combine multiple sources of information (PPI,domains,sequence alignments,etc.) using advanced machine learning algorithms to perform function prediction.These methods have shown improved prediction performance over methods that use only one type of information[29–37].
In this work,we propose a new protein function prediction method,Quantitative Annotation of Unknown STructure (QAUST),which combines the global and local structure similarity search with PPI networks and functional sequence motif detection.Our approach follows a sequenceto-structure-to-function workflow.Starting from the protein amino acid sequence,we first generate structure prediction by the Iterative Threading ASSembly Refinement (ITASSER)method[38].The predicted structure is then used to identify the proteins with similar functions based on a combination of global and local structure similarity search method that follows the same pipeline used in COFACTOR[24,39].PPI information is meanwhile extracted from the STRING database[40].And finally,we extract functionally discriminative sequence motifs as our third main prediction feature.The confidence scores obtained from these three features are combined in a consensus function to obtain our final confidence score.
Since the terminology of a“protein function”might be ambiguous,we would like to clarify that the definitions of function followed in this work are Enzyme Commission(EC)numbers[41]and Gene Ontology(GO)terms[42].EC numbers are used to categorize enzymes into hierarchical families using a numerical classification.Specifically,the EC number(which is composed of four numbers separated by periods,i.e.,A.B.C.D)refers to the reaction catalyzed by a specific enzyme.On the other hand,the GO terms are a set of controlled vocabulary to formally describe proteins and RNAs based upon their functions.Three aspects of ontologies,biological process (BP),cellular component (CC),and molecular function (MF),are defined in this database.Each one of these three GO aspects is represented by a structured directed acyclic graph (DAG),where nodes represent GO terms that describe gene product functions,while the edges represent the relationships (“is_a”or“part_of”) between the GO terms.In GO functional hierarchy,the more general functions are on the top of the graph,while more specific terms are usually present further down the graph.
Our prediction results are compared to the following programs:1)COFACTOR[39],a global and local structure similarity-based method;2) LOMETS [43],a metathreading algorithm;3) HHsearch [5],an HMM-based method that is widely used to detect protein homologs;4)BLAST[3],which transfers annotations based on sequence similarity;5)naïve baseline,which predicts GO terms based on their annotation frequency;and 6) two highly-ranked methods from the Critical Assessment of Functional Annotation (CAFA) assessment [44],GoFDR [32] and INGA [45].
Method
Dataset
To evaluate QAUST for EC prediction,we use the benchmark dataset of COFACTOR[24,39]as our testing dataset.This dataset consists of 318 enzymes with unique EC numbers (first three digits) covering all 6 enzyme classes.Similarly,all sequences in our template libraries having a sequence identity >30% with the query enzymes are excluded from the template libraries.
We evaluate QAUST for GO prediction on a dataset of 500 randomly chosen non-redundant proteins from the CAFA 2 Targets (https://biofunctionprediction.org/cafa/)annotated with at least one GO term.To eliminate any structure or function homologs to the query,templates having a sequence identity >30% with the query proteins are excluded from the template libraries both in the I-TASSER threading library and our function prediction template libraries.
EC number prediction
Global and local similarity search
The first step of our protein function prediction is the generation of the predicted 3D model of the query protein using I-TASSER[38],as outlined in“Section 1”in File S1.The predicted model of the query protein obtained from I-TASSER is then scanned against a non-redundant (pairwise sequence identity no more than 90%) structure template library of 2385 enzymes with at least the first three digits of EC number annotated by the Catalytic Site Atlas(CSA) database [46].This library scanning detects homologous structure templates to the query proteins using two types of structure similarity search programs:global similarity search and local similarity search.
Global similarity search
Templates with a similar global structure to the predicted structure of the query protein are detected from the template library using TM-align [47].Another important consideration when searching for templates with similar global folds to the query protein is the quality of the structural models.Appraising the accuracy of the structure modeling in the scoring scheme helps to reduce the number of false positive predictions.In this particular case,the quality of the predicted I-TASSER model generated in the previous step is evaluated usingCscore[38].
Local similarity search
The local structural search approach consists of three steps(Figure 1).The first step is the structural match of the specific catalytic/active residue pairs.For a given pair of query and template proteins,we first scan the known catalytic/active residues of the template through the query sequence.The query’s residues whose amino acid types are the same as the amino acid types of the template’s catalytic/active residues are marked as potential active sites in the query.The structures of all combined sets of marked residues in the query are extracted from the predicted model and used as candidate active sites.The structures of the candidate sites are superimposed on the known catalytic/active residues in the template.To make the structure superimposition more reliable,for each residuei,the coordinates of Cαatoms and side-chain centers of mass of the two neighboring residues,i.e.,the (i–1)-th and (i+1)-th residues,are also included in the superimposition.

Figure 1 A schematic diagram of the local similarity search procedure for functional site identification
The second step is to identify the key local environment residues around the active sites in the query and the template.For this purpose,we superimpose the complete structure of the query and template proteins based on the rotation matrix obtained from the superimposition of the candidate catalytic/active residue structures obtained in the previous step.A sphere of radiusris then defined around the geometric center of the template’s local 3D fragments,whereris the maximum distance of the template residues in the local 3D fragment from the geometric center.The sphere represents a local environment or a probable active site region,under which the chemical and structural similarity of the query and template are compared.Because a sphere comprising of a very small number of catalytic/active residues can easily generate false positive hits,when the template’s active site region is small,we set the number of residues inside the sphere to be a minimum of 20 residues.This value is obtained using minimum grid search parameter optimization by evaluating different sphere sizes in the range of[10,50]residues to select the most accurate value.
In the third step,the best alignment of the local active site residues in the spheres between the query and the template is identified using a scoring function similar to TM-align.Starting from the initial superposition of the query and template protein structures,we perform a Needleman-Wunsch dynamic programming to generate the best alignment for the residues in the selected sphere of the template and the query,where the alignment score matrixSijfor aligning thei-th residue in the query and thej-th residue in the template is defined as:

wheredijis the Cαdistance between residuesiandj,d0is the distance cutoff given byd0=1.8obtained from TM-align,Mijis the substitution score between thei-th andj-th residues taken from the BLOSUM62 mutation matrix with the value normalized by the diagonal element in the mutation matrix.The gap penalty is set as −1.For a given scoring matrixSij,a new alignment is generated by dynamic programming.A new superposition and scoring matrix are then constructed based on the new alignment to obtain a newer alignment from dynamic programming.This procedure is iteratively repeated until the final alignment is converged.For each alignment,the active site match(AcM)is evaluated using an alignment score defined as:

whereNtrepresents the number of residues in the active site sphere of the template,Naliis the number of aligned residue pairs.The maximum AcM score obtained during the heuristic iterations is recorded for each candidate active site.Finally,the set of residues in the candidate active site which has the highest AcM score is selected to evaluate the similarity between the active sites of the query and the template.The weights that form the AcM score have been derived based on the predicted structures of 100 randomly chosen training proteins from the template library,which are non-homologous(sequence similarity<30%)to the test proteins in order to maximize the sensitivity and specificity of the predictions.
Scoring function for global and local similarity search
The final score for predicting EC numbers,used to sort the hits from the enzyme library,is a combination of the global similarity search score and the AcM score (obtained from the local similarity search)and is defined as:
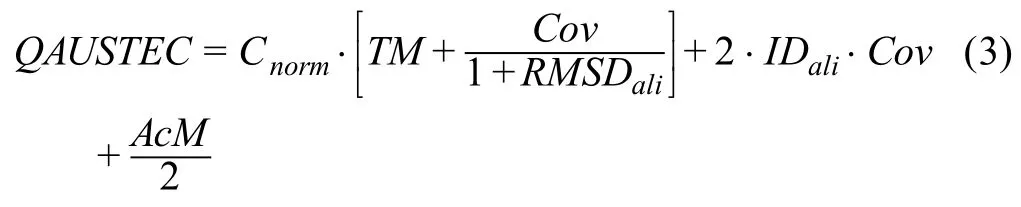
whereCovrepresents the coverage of the structural alignment,RMSDaliis the root-mean-square deviation (RMSD)between the model and the template structure in the structurally aligned region,andIDaliis the sequence identity between query and template based on the alignment generated by TM-align.The hyperbolic-tangent-like normalization is further used to normalize the raw EC score to be between 0 and 1:

GO term prediction
For GO term prediction,we combine three different predictors.Each one of these predictors generates a confidence score.The three confidence scores obtained are then combined in a consensus function to generate the final prediction score.The first predictor is the global structure similarity,which uses I-TASSER to predict the 3D structure of the query,and then scans a library of templates to identify those which have a similar global structure to the predicted model.The second predictor is based on PPI information,and the third one is based on extracted functional sequence motifs.
Global protein structure similarity
Similar to EC prediction,I-TASSER is also used here to construct the corresponding 3D model to the query sequence.The model obtained is then scanned against a library of templates to identify those which share a similar global structure to the query model (https://zhanglab.ccmb.med.umich.edu/BioLiP/library.html).For the time being,the functionally important residues for most of the proteins in the GO template library are unknown.Therefore,only the global similarity search is taken into consideration when sorting the hits from the GO library.Global similarity search for GO prediction is done in a similar way to global similarity search for EC prediction described in the previous section.The only difference is that to select the best hits for GO prediction,we rank a template using theFhscoredefined as:
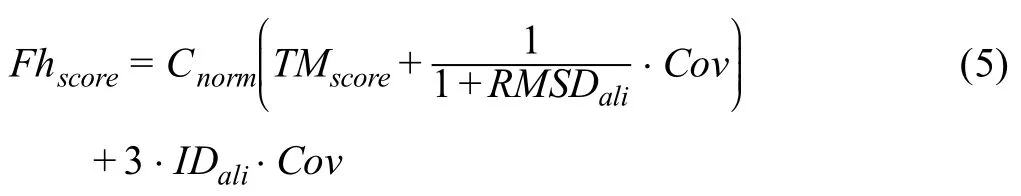
Since each single protein can be annotated with multiple GO terms and the global search may result in many close template structures,a query protein can have multiple GO term predictions with highFhscore.Therefore,the confidence score of each GO term is calculated as follows:

whereλrepresents a given GO term,Nλis the number of templates annotated with the GO termλ,andNis the total number of templates selected for generating the consensus.When multiple close templates are available,we only consider the templates withFhscore>1.For those query proteins with less than 10 templates ofFhscore>1,the top 10 templates are selected for generating the consensus prediction regardless of theFhscore.Also,given the hierarchical nature of the GO DAG,we consider that when a protein is annotated with a given GO term,all its ancestor GO terms(through“is_a”relation) are automatically implied.Therefore,once a GO termλis scored,we score all its ancestor terms as well.The score of any ancestor GO termμof termλis calculated as:
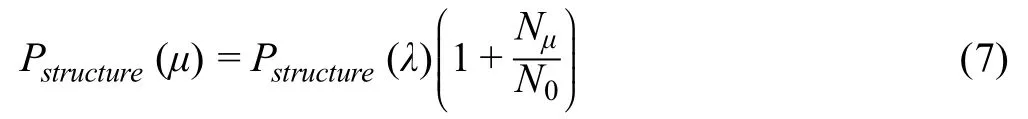
whereNμandN0are the numbers of leaf nodes under nodeμand the root node,respectively.Since COFACTOR[24,39]uses a similar structure scoring function,we have highlighted the main differences between QAUST and COFACTOR in“Section 2”in File S1,and compared our method with COFACTOR in the experiments.
PPI network
We exploit the information provided by the STRING [40]database,which is a library of PPI networks,to extend our prediction set.The query protein sequence is mapped to its corresponding STRING entry by BLAST,with minimum sequence identity cutoff of 90%.Extracting the PPI partners of the query,we calculate the confidence score of STRING for a GO termλas the frequency of the GO termλamong the experimentally annotated interaction partners of the query protein:

whereis the number of interaction partners annotated with the GO term,andNis the number of partners associated with term,according to the corresponding UniProt-GOA (http://www.ebi.ac.uk/GOA) entry of this PPI partner.This score could take any value from 0 to 1.
Functionally discriminative sequence motifs
In addition to the structure similarity search and PPI features discussed above,we also include sequentially extracted features to predict GO terms since a sequence is a highly valuable source of information that can especially be useful when dealing with proteins for which we cannot construct a good quality 3D structure model or which with no known PPI information.
Our functionally discriminative motif detection algorithm follows three steps:detection of sequence templates,identification of functionally discriminative motifs given a GO term,and scoring the query protein.
Detection of sequence templates for a query protein
The sequence homologs of a query sequence are detected by PSI-BLAST[3]from the Uniref90 database[49].We filter all obtained homologs having a sequence identity >30%with the query.
Identification of functionally discriminative motifs given a GO term
We map all the selected sequence homologs of the query to their corresponding GO annotations in the UniProt-GOA database (http://www.ebi.ac.uk/GOA).GO terms assigned with“Inferred from Electronic Annotation (IEA)”or“No biological Data available (ND)”evidence codes are not considered.We also filter out annotations with the evidence code“Inferred from Physical Interactions (IPI)”,since we use PPI information in our features.After filtering these annotations,we are left with the annotations based on evidence codes:Inferred from Experiment (EXP),Inferred from Direct Assay(IDA),Inferred from Mutant Phenotype(IMP),Inferred from Genetic Interaction (IGI),Inferred from Expression Pattern(IEP),Traceable Author Statement(TAS),and Inferred by Curator (IC).For each GO termλ,we build two sets of sequences from the set of homolog sequences detected in the previous step.These two sets are:the“annotated set”,which is the set of sequence homologs annotated with this specific GO term,and the“notannotated set”,which is the set of sequence homologs not annotated with this given GO term.For each of these two sets,we extract the ten most frequent motifs by extracting all unique amino acid motifs of length [4,7] from the sequence set using sliding windows.These motifs are ranked in descending order by their occurrences.The top 10 most frequent motifs are the initial“frequent list”,while the remaining motifs are in the“waiting list”.If,within the“frequent list”,a short motif is a substring of another longer motif,the shorter motif is discarded,and the most frequent motif from the“waiting list”is transferred to“frequent list”to ensure that the latter always has 10 motifs.This process is iterated until,in the“frequent list”,any motif is not a substring of another motif.The motifs in the“frequent list”are used for matching the query in the next step.
Scoring the query protein
For each of the two sets(annotated and not-annotated sets),we check the number of frequent motifs extracted in the previous step that are also present in the query sequence.Then,we calculate the confidence score of the GO term given the query sequence as follows:

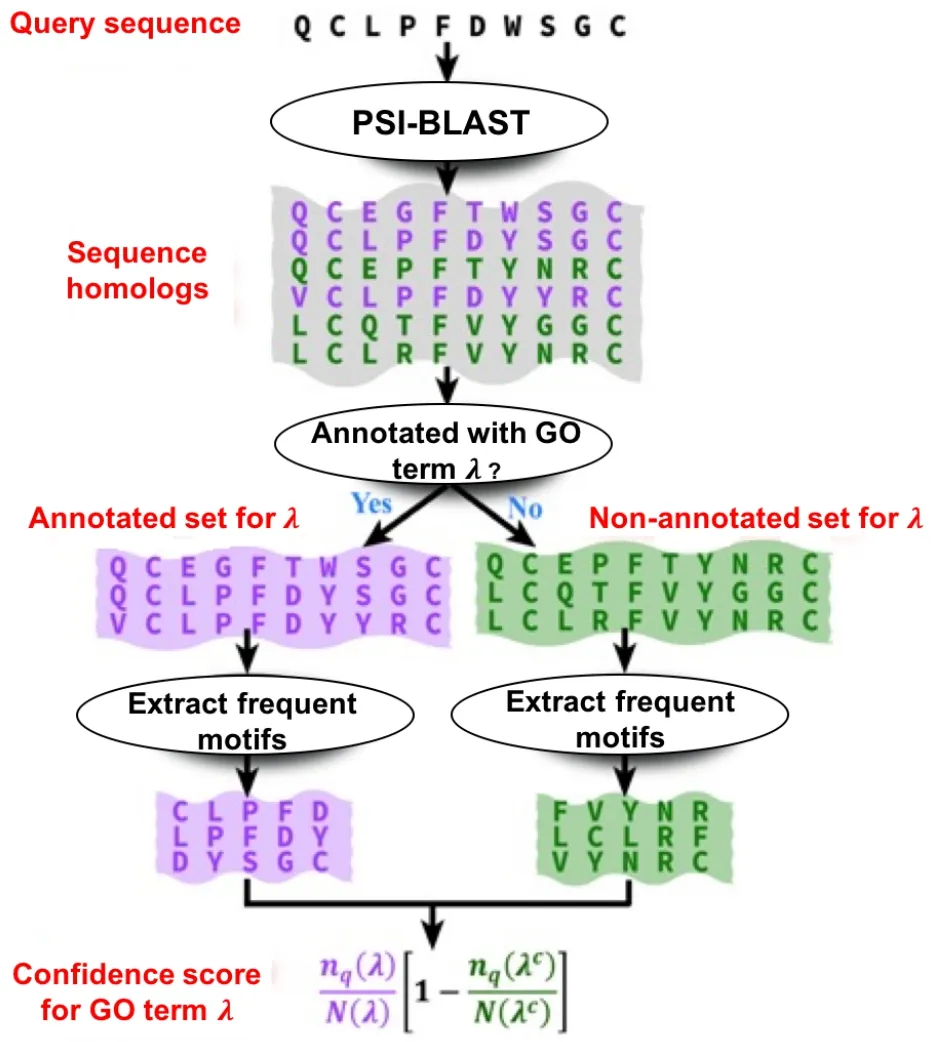
Figure 2 Workflow for sequence motif-based function prediction in QAUST
Consensus
To predict GO terms,the three main scores obtained from the three different predictors (the structure search,the PPI network,and the functional motifs) are combined by consensus averaging to calculate the final confidence score

This equation used to calculate the consensus has beenpreviously used by other methods for protein function prediction [45].If one or more predictors are not available for a given term(e.g.,no interaction partners are known for the given query),only the available predictors are used to obtain the confidence score.Also,since GO uses the truepath rule(i.e.,if a protein is associated by a term,it is also implicitly annotated by its ancestors),for every predicted GO term,all its ancestors are considered to be predicted as well since they are more general terms.
Cell culture,plasmid construction,and transfection
The Hek293T cells were obtained from the American Type Culture Collection (ATCC),and were cultured in DMEM(Catalog No.C11965500BT,Gibco) supplemented with 10% fetal bovine serum (FBS;Catalog No.10270106,Gibco) and 1% penicillin/streptomycin (P/S;Catalog No.15070063,Gibco) with 5%CO2at 37°C.
To establish the constructs expressing human tripartite motif-containing 22 (TRIM22) protein,we cloned the coding sequence of humanTRIM22withFLAGorGFPat its N-terminus into pcDNA3.1(+) vector,using one-step clone kit(Vazyme).
For transfection,PEI reagent was applied with the general ratio of 30 reagents as 10 μg plasmids (5 μgFLAGTRIM22and 5 μgGFP-TRIM22) into each 10 cm plate at 70% cell confluence.After 48 h transfection,cells were harvested for the following assays.
Co-immunoprecipitation
Cells were harvested with lysis buffer (20 mM Tris-HCl pH7.5,150 mM NaCl,1 mM EDTA,1% NP-40) with proteinase inhibitor.For each sample,25 μl protein A/G beads (MCE) were incubated with 1 μg anti-FLAG (Sigma) or anti-GFP (Proteintech) antibody at 4°C for 2 h.Mouse IgG (Cell Signaling Technology) was used as a negative control.Beads were washed three times with lysis buffer and then incubated with 500 μg cell lysate at 4°C for another 2 h.After washed with lysis buffer three times,20 μl 2×sample buffer were added into beads and heated at 95°C for 10 min.Protein levels of beads or cell lysate were then detected by Western blot using individual antibodies.
Results
Prediction of enzyme functions (EC numbers)
We compared the EC prediction performance of our method to five other methods:HHsearch [5],LOMETS [43],BLAST [3],COFACTOR [24,39],and DEEPre webserver[49].We compared the performance of these methods based on precision (positive predictive value) and recall (sensitivity) rates.Figure 3shows the precision-recall graph corresponding to four baseline methods as well as QAUST.Since the DEEPre webserver does not report the confidence score with the annotation,we could not draw the precisionrecall curve but compared QAUST to DEEPre based on accuracy.An EC number prediction is considered to be“true”if the first three digits of the EC number from the hit are identical to those of the query protein;otherwise the hit is considered to be“false”.As shown in Figure 3,the rate of true positive predictions using the EC-score is much higher than that of HHsearch,LOMETS,BLAST,and COFACTOR at most recall rates.QAUST has also an area under precision-recall curve (AUPRC) of 0.712 which is higher than that of COFACTOR(0.643),LOMETS(0.510),and HHsearch (0.489).Table 1reports the accuracy of QAUST compared to five other methods.The results show that DEEPre has a slightly higher performance than QAUST in terms of accuracy,which is probably due to the fact that DEEPre is a machine learning method trained on a large number of enzymes with known functions that overlap or contain close homologs to our test data.
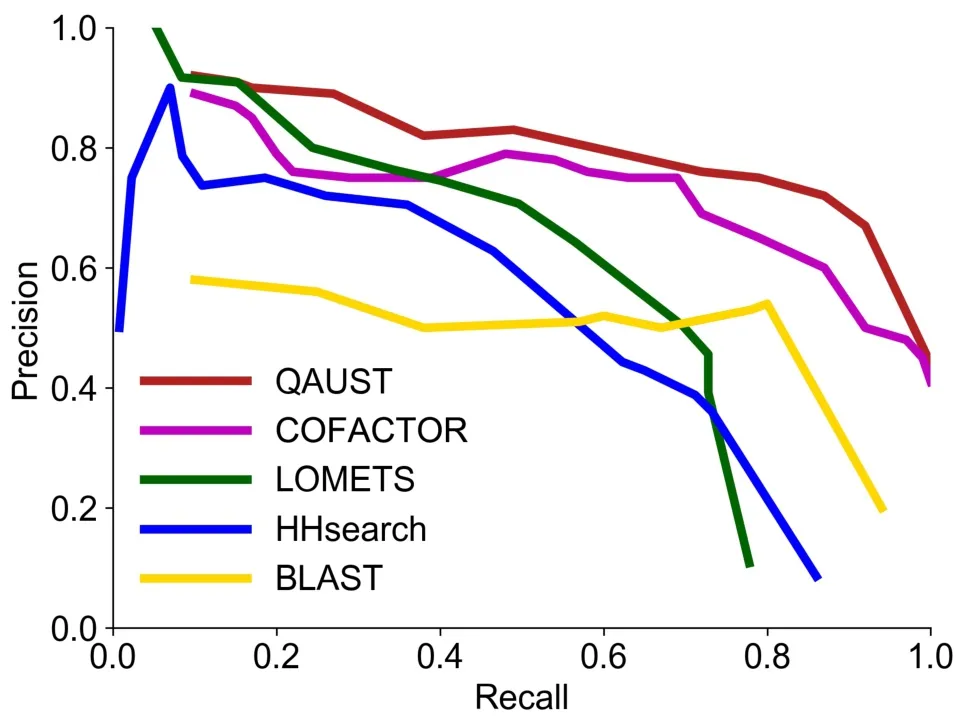
Figure 3 Precision-recall curves for EC prediction by QAUST,COFACTOR,LOMETS,HHsearch,and BLAST
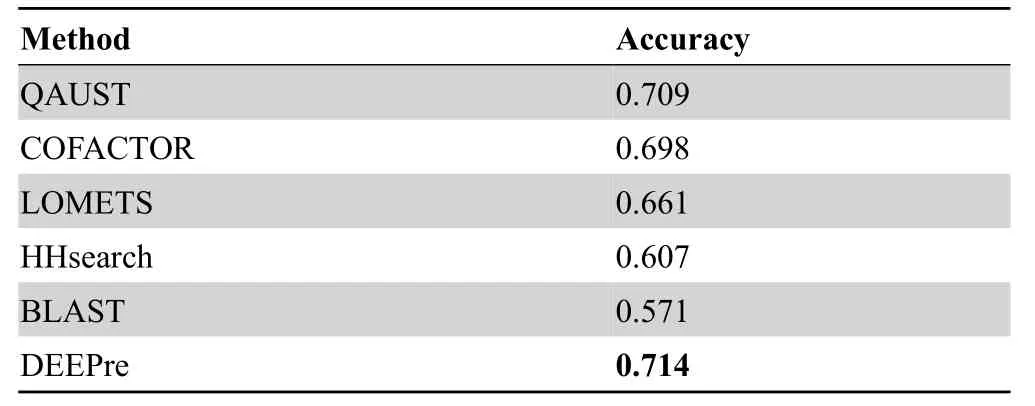
Table 1 Accuracy values of EC prediction for QAUST and five other methods
Prediction of GO terms
To assess the contribution of individual predictors to the GO prediction performance by QAUST,we visualized the precisionrecall curve of the structure similarity search alone(Pstructure;corresponding to the COFACTOR mothed[39]),the precisionrecall curve of structure similarity search combined with PPI information(PstructureandPSTRING),and that of the final QAUST prediction (Pstructure,PSTRING,andPMOTIF).Additionally,we compared the prediction performances of our method based on different sets of features on our dataset(see the subsection Dataset in Method) to those of naïve baseline (a method predicting GO terms based on their annotation frequency),BLAST [3],LOMETS [43],HHsearch [5],INGA webserver [45] (a method combining BLAST,PPI information,and Pfam in one predictor),and GoFDR[32](one of the top function prediction methods at the CAFA assessment [44] which uses a machine learning model as classifier and discriminative residues as the main feature).
The performance was primarily evaluated using precision-recall curves computed at each prediction score threshold.We also used the Fmaxmeasure as a quantitative measure to evaluate the overall performance of the precision-recall curves.Precision,recall,and Fmaxare defined in the same way as the CAFA evaluation[50].The Fmaxmeasure is computed as the maximum value of the Fmeasurewhich is computed at each threshold as

As shown inFigure 4,our method combining structure,PPI,and functional motif information achieves higher precision than most other methods at most recall points,in particular for BP and CC.For our dataset,structure and motif information has been used for all proteins.However,the PPI information from STRING is missing for 74 proteins.In this case,only the structure and motif information are used.The Fmaxmeasure values are shown inTable 2,and the Sminvalues are shown inTable 3.Surprisingly,in CC prediction,naïve baseline,which predicts GO terms based on their annotation frequency,achieves higher performance than all other methods including QAUST.In fact,in the CAFA assessment [44],naïve baseline also outperforms most of the other methods in predicting CC terms.One possible explanation for why naïve baseline has higher Fmaxfor CC term prediction is because the most frequently used CC terms in protein annotation are usually part of a small set of very general terms such as“cytoplasm”or“intracellular part”.Since the naïve baseline is solely based on frequency,it increases the chance of predicting a true positive[44].We also reported thePvalues obtained from the Mann-WhitneyUtest to assess the significance of the difference in performance of QAUST compared to all other methods in“Section 3”in File S1 and Table S1.
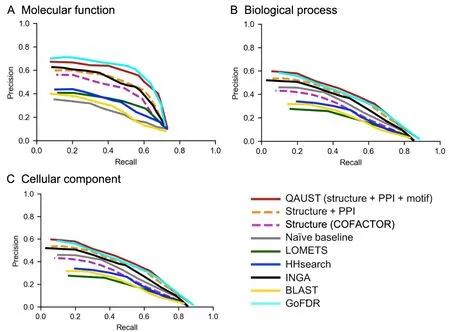
Figure 4 Precision-recall curves for GO prediction
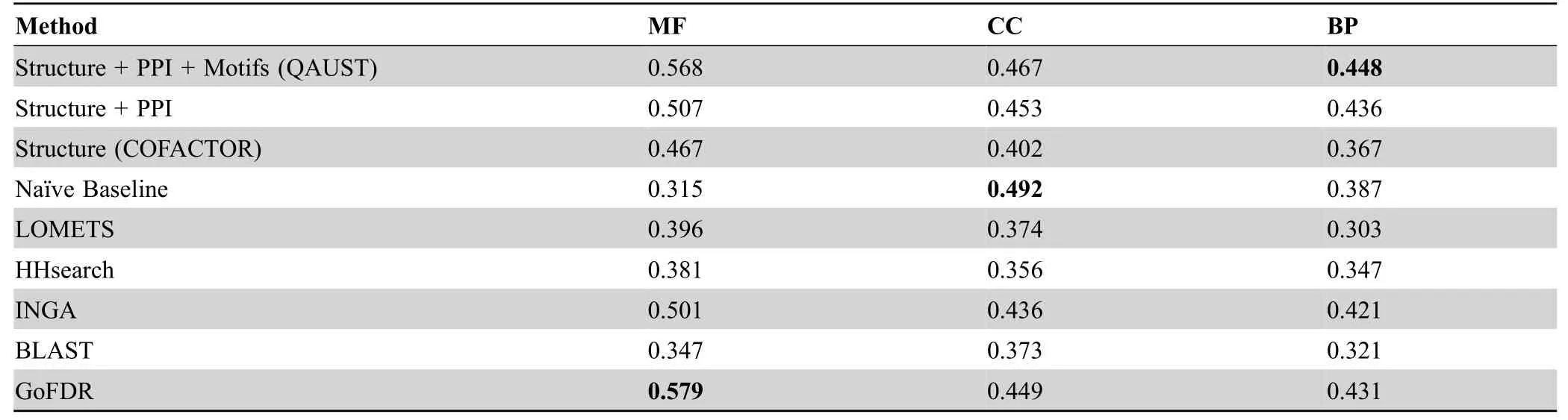
Note:The highest Fmax value of each branch of GO is shown in bold.GO,Gene Ontology;MF,molecular function;CC,cellular component;BP,biological process.
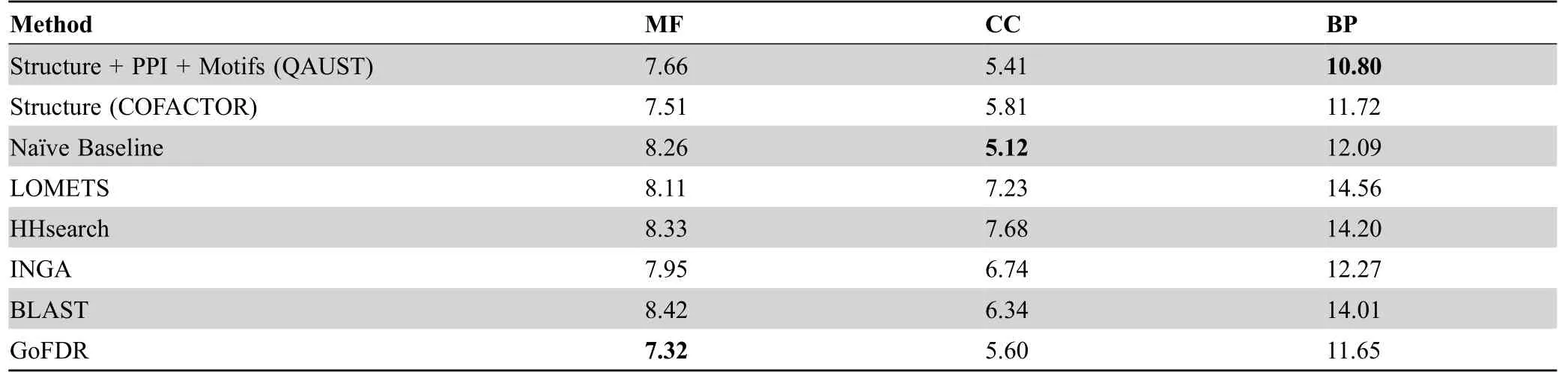
Note:The highest Smin value of each branch of GO is shown in bold.Smin,minimum semantic distance.
As a further analysis,to investigate if the performance of our method is solely due to the power of the I-TASSER structure prediction we used,we replaced the I-TASSER structure prediction component of our method by HHsearch and LOMETS structure prediction,respectively.Our results show that no matter which structure prediction method is used,our scoring function,Pstructure,can significantly improve the performance on predicting the GO terms.Meanwhile,among the three structure prediction methods,I-TASSER withPstructureconsistently performs the best over all three GO hierarchy branches of MF,BP,and CC,whereas LOMETS withPstructurehas the second best performance on CC,and HHsearch withPstructureis the second best on predicting MF and BP terms (File S1,Section 4;Figure S1).Additionally,we evaluated the performance of our method when only PPI and motif information are used without including any structure-based information.The results show that the function prediction performance drops when structure features are not used (File S1,Section 5;Figure S2).
How do PPI information and functional sequence motifs improve the prediction?
PPI information extracted from STRING is an important feature used in our prediction.In Figure 4,we show how PPI information alone improves the performance achieved by the structure similarity search(orange dash linesversusmagenta dash lines).The precision-recall curves in Figure 4 show that the contribution of PPI information from STRING is very significant for CC and BP terms,especially for large recall rates.Moreover,the precision-recall curves confirm our initial hypothesis on the utility of PPI information for function annotation.As shown in Figure 4,while there is some improvement in predicting MF terms,this improvement is not substantial.The reason why PPI is not particularly helpful in MF term prediction is most probably because proteins that interact with each other do not necessarily share the same specific molecular function,even when they are part of the same biological process.
In addition to the structure similarity search and the PPI features,the results show that the functional motifs extracted improve the performance of the prediction significantly.As a further analysis,we evaluated the performance of our functional motif detection method when both predicted and experimentally annotated GO terms are taken into consideration instead of considering experimental annotations only.The results of this experiment are reported in“Section 6”in File S1 and Table S2.In addition to comparing the performance of our method to BLAST,LOMETS,HHsearch,and COFACTOR,we also compared it to INGA and GoFDR,two top methods from CAFA [44] in particular for MF and BP term prediction,and to naïve baseline which is one of the performance references used in CAFA.
Case studies
Prediction of the bacteriophage T4 gene 59 helicase assembly protein
To better illustrate the performance of QAUST and the contribution of each component to the prediction,we used an example bacteriophage T4 gene 59 helicase assembly protein(PDB ID P13342;the cyan structure inFigure 5A),which is a DNA binding protein required mainly for DNA replication in the late stage of T4 infection[51].Figure 5B shows the set of BP terms associated with this protein.In this particular example,both BLAST and INGA did not predict any correct term for this protein(the naïve root term is not counted here).When solely using global structure similarity (Pstructure),we could only predict one single correct BP term.This makes sense because all the queries in our test set are difficult targets,which do not have close homologs in the template database.For instance,the closest template for this query P13342 is the methionine-tRNA ligase (PDB ID 2CT8A;the magenta structure in Figure 5A).The sequence identity between P13342 and 2CT8A is only 6.84%and the TM-score between the two structures is only 0.24.Therefore,structure similarity or homologybased methods are not expected to predict the function of the query well.Structure information (Pstructure) combined with PPI predicted three correct terms out of the six experimentally annotated terms.On the other hand,QAUST predicted four correct terms out of six.In addition,the prediction of QAUST is at least one level deeper in the GO hierarchy than the other methods.Meanwhile,the predicted MF and CC terms for this protein by QAUST are at least as accurate as other methods.Two other case studies illustrating examples for predicting MF and CC terms are also shown(File S1,Section 7 and Section 8;Figures S3 and S4).

Figure 5 A study case for protein function prediction using QAUST
Experimental validation of TRIM22 dimerization
To provide an experimental assessment of the performance of QAUST,we chosed the human TRIM22 protein as an example.TRIM22 is known as an interferon-inducible protein which shows antiviral activity,such as HIV,HBV,and HCV [52–54].Recent studies have also shown that TRIM22 mediates autophagy in human macrophages [55].However,the function of TRIM22 is still not comprehensively understood as the protein only exists in primates.
We applied QAUST to predict the function for TRIM22.Among the predicted GO terms with high consensus scores(File S1,Section 9;Table S3),some of the CC and BP terms agree well with the previously known functions of TRIM22,such as the CC term“nucleus”and the BP term“response to virus”.However,the only two predicted MF terms have quite high consensus scores,“protein binding”and“protein homodimerization activity”,suggesting that TRIM22 binds to itself to form a dimer.
We thus set out to test the binding ability of human TRIM22 using co-immunoprecipitation(Co-IP)(Figure 6A).First,FLAG-tagged and GFP-tagged human TRIM22 proteins were co-expressed in HEK293T cells(Figure 6B).To prove their interaction,we then pulled down FLAGtagged TRIM22 from the cell lysate.Western blot showed that when FLAG-tagged TRIM22 was pulled down,GFP-tagged TRIM22 can be detected by anti-GFP antibody(Figure 6C),suggesting that GFP-tagged and FLAG-tagged TRIM22 proteins exist in the same complex in HEK293T cells.To further confirm this binding,we did Co-IP in the opposite way.As expected,FLAG-tagged TRIM22 was also detected in IP of GFP-tagged TRIM22 (Figure 6D).Taken together,our results detected multiple TRIM22 proteins in the same complex,which provides the possibility of its binding with each other.
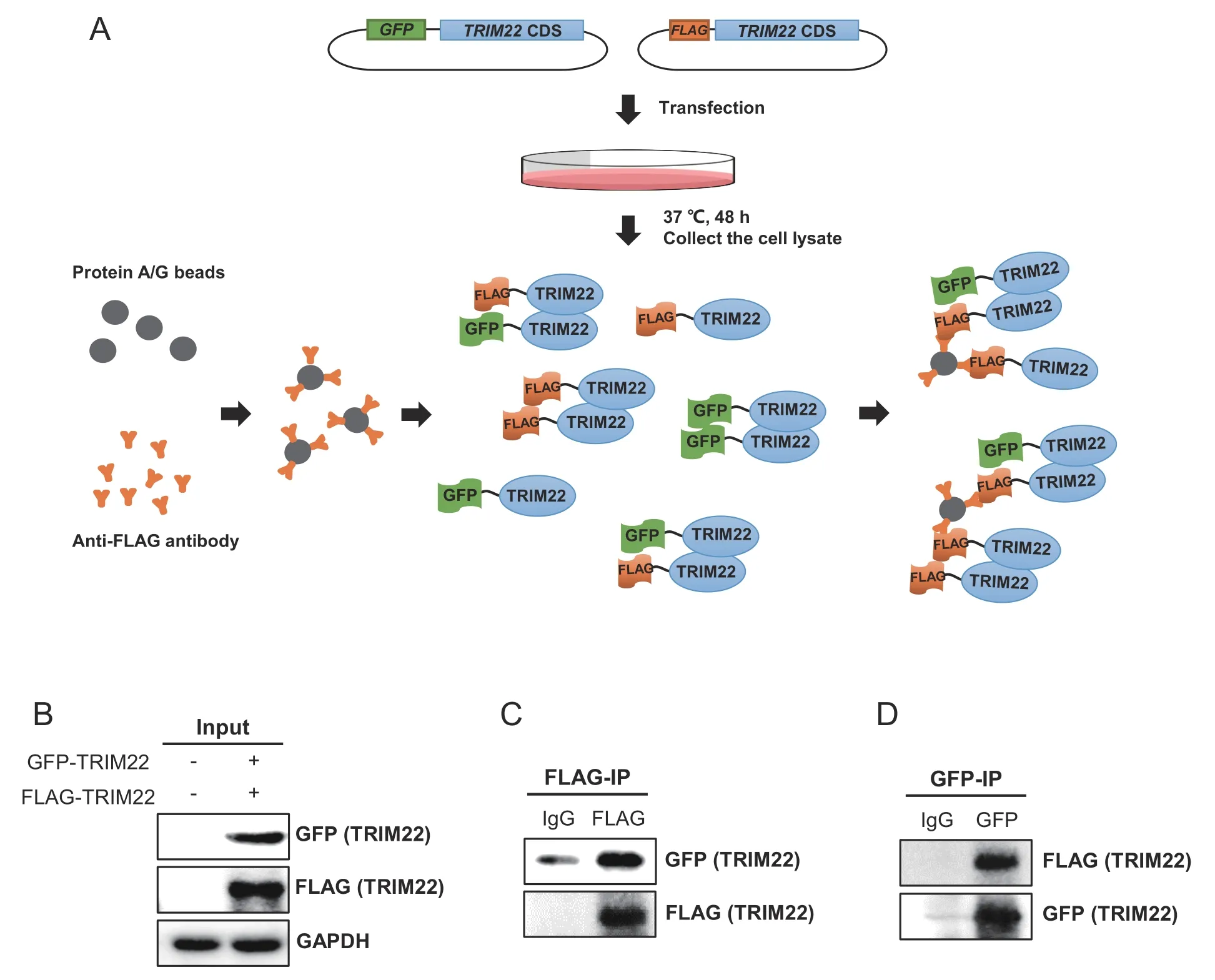
Figure 6 Experimental validation of homodimerization function of human TRIM22
Discussion
In this work,we develop QAUST,a method to predict biological functions of protein molecules using three main features:global and local protein structure similarity,PPI information,and functional sequence motifs.In our method,we construct the 3D structure from the amino acid sequence using I-TASSER.Functional analogs are then identified by performing global and local structural similarity search through the functional libraries,with the scoring function involving the confidence score of structural predictions,sequence and structural similarity of the I-TASSER model with the functional templates,and the local active site matches.We have also tried to improve the performance of GO prediction by incorporating PPI information,especially in order to improve the prediction of GO terms under BP and CC aspects.We further developed a novel predictor that extracts functional motifs that are related to a specific GO term and used it as our third predictor.
On a set of 500 non-redundant proteins,QAUST is shown to have higher function prediction accuracy than the other competing methods on most prediction tasks.This performance advantage is mainly a result of combining three different predictors which cover major aspects of proteins.Additionally,our three prediction components complement each other in the sense that they contribute differently to the prediction of the three aspects of GO.While PPI information improves significantly the prediction of BP and CC terms,functional motif detection is mainly useful in improving MF term prediction.However,QAUST has a number of limitations that give room for possible improvement in the future.One main limitation is that QAUST is much more expensive in terms of running time compared to the other methods as reported in“Section 10”in File S1 and Table S4.The second limitation is that our method cannot be directly used to infer functions that are not included in EC or GO systems,since it solely infers protein functions from existing protein annotations.Finally,given that the three components we used work differently in predicting different aspects of GO,it may be helpful to weight their scores differently depending on the nature of the GO term evaluated instead of combining the scores in a simple consensus.In particular,advanced machine learning methods,such as deep learning[56–60],could help weight and combine the scores in a more efficient way to obtain better prediction results which could be a possible future improvement of this work.
Code availability
QAUST can be accessed at http://www.cbrc.kaust.edu.sa/qaust/submit/.
CRediT author statement
Fatima Zohra Smaili:Conceptualization,Methodology,Validation,Writing -original draft.Ambrish Roy:Writing -original draft,Data curation.Meshari Alazmi:Software.Stefan T.Arold:Writing -review &editing.Srayanta Mukherjee:Writing -original draft.P.Scott Hefty:Writing -original draft.Shuye Tian:Validation,Writing-original draft.Wei Chen:Validation,Supervision.Xin Gao:Conceptualization,Methodology,Validation,Writing -review &editing,Supervision.All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interest.
Acknowledgments
We thank Mr.Chengxin Zhang,Dr.Wei Zhang,and Prof.Yang Zhang for helpful discussions.This work was supported by the King Abdullah University of Science and Technology(KAUST)Office of Sponsored Research(OSR)(Grant Nos.URF/1/1976-04 and URF/1/1976-06).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.02.001.
ORCID
0000-0001-6439-0659 (Fatima Zohra Smaili)
0000-0002-1832-5765 (Shuye Tian)
0000-0003-1738-0591 (Ambrish Roy)
0000-0001-9074-1029 (Meshari Alazmi)
0000-0001-5278-0668 (Stefan T.Arold)
0000-0002-2750-5071 (Srayanta Mukherjee)
0000-0002-2303-2465 (P.Scott Hefty)
0000-0003-3263-1627 (Wei Chen)
0000-0002-7108-3574 (Xin Gao)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- rePROBE:Workflow for Revised Probe Assignment and Updated Probe-set Annotation in Microarrays
- BrcaSeg:A Deep Learning Approach for Tissue Quantification and Genomic Correlations of Histopathological Images
- TSUNAMI:Translational Bioinformatics Tool Suite for Network Analysis and Mining
- Computational Assessment of Protein–protein Binding Affinity by Reversely Engineering the Energetics in Protein Complexes
- Identifying Novel Drug Targets by iDTPnd:A Case Study of Kinase Inhibitors
- eTumorMetastasis:A Network-based Algorithm Predicts Clinical Outcomes Using Whole-exome Sequencing Data of Cancer Patients