弥勒褐煤结构特征及其分子模型构建
2021-07-29张殿凯李艳红常丽萍訾昌毓张远琴田国才赵文波
张殿凯 ,李艳红,,* ,常丽萍 ,訾昌毓 ,张远琴 ,田国才 ,赵文波
(1. 昆明理工大学 化学工程学院,云南 昆明 650500;2. 太原理工大学 煤科学与技术省部共建国家重点实验室培育基地,山西 太原 030024;3. 昆明理工大学 省部共建复杂有色金属资源清洁利用国家重点实验室,云南 昆明 650093)
中国褐煤资源丰富,但因其具有热值低、易风化、易自燃和高含水量的特性,一般仅用作低级燃料进行燃烧,这不仅污染了环境,也造成了资源的大量浪费[1]。研究褐煤的分子结构,是探究褐煤理化性质和反应活性的前提,也是褐煤高附加值利用的基础[2-5]。
煤结构模型的构建是从分子水平上研究煤的性质。煤结构的参数信息可利用先进的现代分析手段来获取,主要包括13C 固体核磁共振(13C NMR)、红外光谱(FT-IR)、X射线光电子能谱(XPS)、X射线衍射(XRD)等[6]。葛涛等[7]联用FT-IR、XPS和13C NMR技术,研究了高阳炼焦煤中的碳和氧在煤分子中的存在形式,并提出了含碳、含氧结构的定量和定性分析方法。Li等[8]采用多种现代分析技术研究了昭通褐煤的结构特性,并对煤中氮的存在形式及其乙醇提取物中的有机氮化合物的分子组成进行研究。张帅等[9]基于HRTEM、XPS、13C NMR等分析结果,对煤分子结构进行研究,成功构建出内蒙古扎鲁特地区无烟煤分子结构模型。随着计算机技术的发展,可以通过计算机辅助分子设计的方法构建煤分子模型。对此,已有相关学者展开了研究[10-13]。由于褐煤的复杂性、多样性以及异质性的特点,褐煤分子结构的研究相对滞后[2]。目前的研究主要集中于褐煤分子结构特性规律的探索,而关于褐煤分子结构模型的建立研究报道却很少。
本研究以云南弥勒褐煤为研究对象,利用元素分析、13C NMR、FT-IR和XPS等现代分析技术对煤结构特性进行研究,并以此为基础构建出弥勒褐煤的分子平均结构模型。采用密度泛函理论、半经验和从头算的方法,对分子模型进行构型优化与光谱模拟。最后,通过对比模拟FT-IR谱图、模拟13C NMR谱图与实验谱图的吻合度,验证所构建的分子模型的准确性。
1 实验部分
1.1 煤质分析
煤样来自于云南省弥勒市,实验前将样品粉碎、筛分至粒径小于74 μm,置于105-110 ℃干燥箱中,在氮气流中干燥至恒重,取出放于无水、氮气气氛下保存待用。弥勒褐煤(ML)的工业分析、元素分析见表1。其中,煤样的工业分析依据国家标准GB /T 212—2008测定。煤样元素分析中,依据GB/T 476—2008测定碳(C)、氢(H)含量,依据GB/T 19227—2008测定氮(N)含量、依据GB/T 214—2007测定全硫(S)含量,最后采用差减法获得氧(O)含量。
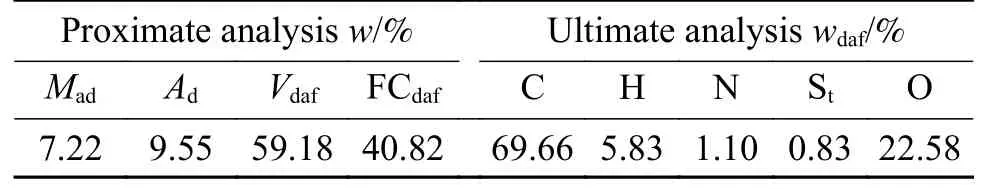
表1 ML的工业分析与元素分析Table 1 Proximate analysis and Ultimate analysis of ML
1.2 13C固体核磁共振波谱分析
采用德国布鲁克公司生产的Bruker AVWB III600交叉极化魔角核磁共振波谱仪(13C CP/MAS NMR)对褐煤进行分析。测试条件:共振频率为150.9 MHz,魔角旋转速为12 kHz,接触时间为4 ms,脉冲延迟为3 s,13C的化学位移采用TMS定标。
1.3 傅里叶红外光谱分析
采用德国布鲁克(Bruker)ALPHA傅里叶红外光谱分析仪对原煤的官能团进行分析。测定条件:KBr压片法(2∶120),400-4000 cm-1扫描,扫描光谱分辨率为4 cm-1。
1.4 X射线光电子能谱分析
采用多功能扫描成像光电子谱(PHI5000 Versaprobe-II 附加扫描俄歇电子能谱)进行表面元素分析。使用X光源为单色光的AlKα(hD=1486.6 eV)源,功率50 W,电压15 kV,使用C 1s284.8 eV校准。
1.5 模型构建步骤
分子模型的构建主要分为结构特性分析和量子化学计算两大部分,构建流程如图1所示。
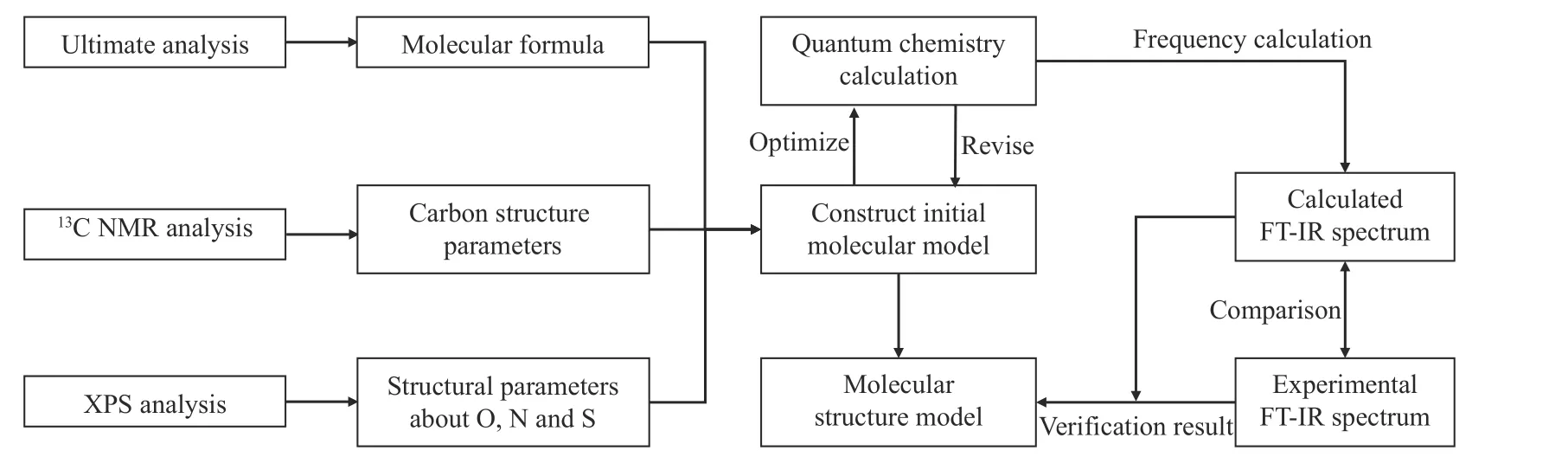
图1 分子模型构建流程图Figure 1 Procedure of model construction
1.6 分子结构模型优化
借助Gaussian 09软件对ML分子模型进行构型优化和振动频率分析[14]。构型优化采用半经验法PM3和密度泛函理论框架下的全电子M06-2X泛函[15];采用从头算HF/3-21G基组计算分子结构模型的模拟FT-IR谱图;采用规范无关原子轨道(gauge-independant atomic orbital, GIAO)方法GIAO/6-31G模拟13C NMR谱图。
2 结果与讨论
2.1 13C CP /MAS NMR核磁共振波谱分析
在褐煤的核磁共振波谱中,主要存在三个化学区域:脂肪部分(0-90)、芳香部分(90-170)以及羰基部分(170-220)。芳香碳部分主要由质子化芳香碳和非质子化芳香碳组成。其中,非质子化芳香碳主要包括芳香桥头碳、烷基取代芳香碳以及芳香酚。脂肪碳部分主要由甲基碳、亚甲基碳、季碳以及含氧脂肪碳组成[8,16,17]。如图2所示,使用origin 2018软件将波谱分为18个峰,分别代表不同的碳类型。其中,13C NMR峰位和化学位移(δ)归属参考了相关文献[18-21]。
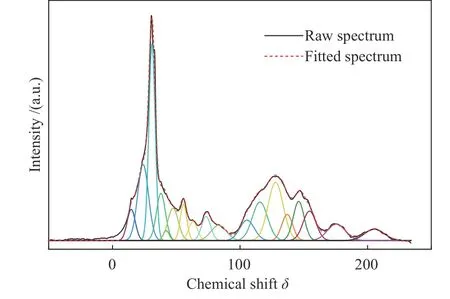
图2 ML的 13C NMR分峰拟合谱图Figure 2 13C NMR peak fitting spectra of ML
从图2中可以看出,峰主要集中在0-170,这表明了ML大分子碳骨架主要为脂肪族碳和芳香族碳。在脂肪族碳区域,以δ为31处的主峰代表了亚甲基碳,δ为24处的肩峰源自芳环上的甲基。此外,在47-90的谱带中出现一些相对强烈的共振信号,这些信号来自与氧结合的脂肪族碳。在芳香族碳中,含量最多的是质子化芳香碳,其次是烷基取代芳香碳。结合表2所列数据,通过计算可得ML各项参数:芳碳率(fa=fao1+fao2+faH+fab+fas)为38.79%;脂碳率(fal=fal1+fala+fal2+fal3+falo)为54.18%;羧基碳含量(faC=faC1+faC2)为7.03%;芳香桥头碳/芳香周边碳(χb=fab/(faH+fas+fao2))为0.11;平均亚甲基链长(Cn=fal2/fas)为3.8;芳香环取代度(σ=(faH+faS+fao2)/fa)为75%,即苯环平均取代基数量为3。以上参数均为构建ML结构模型的重要依据。
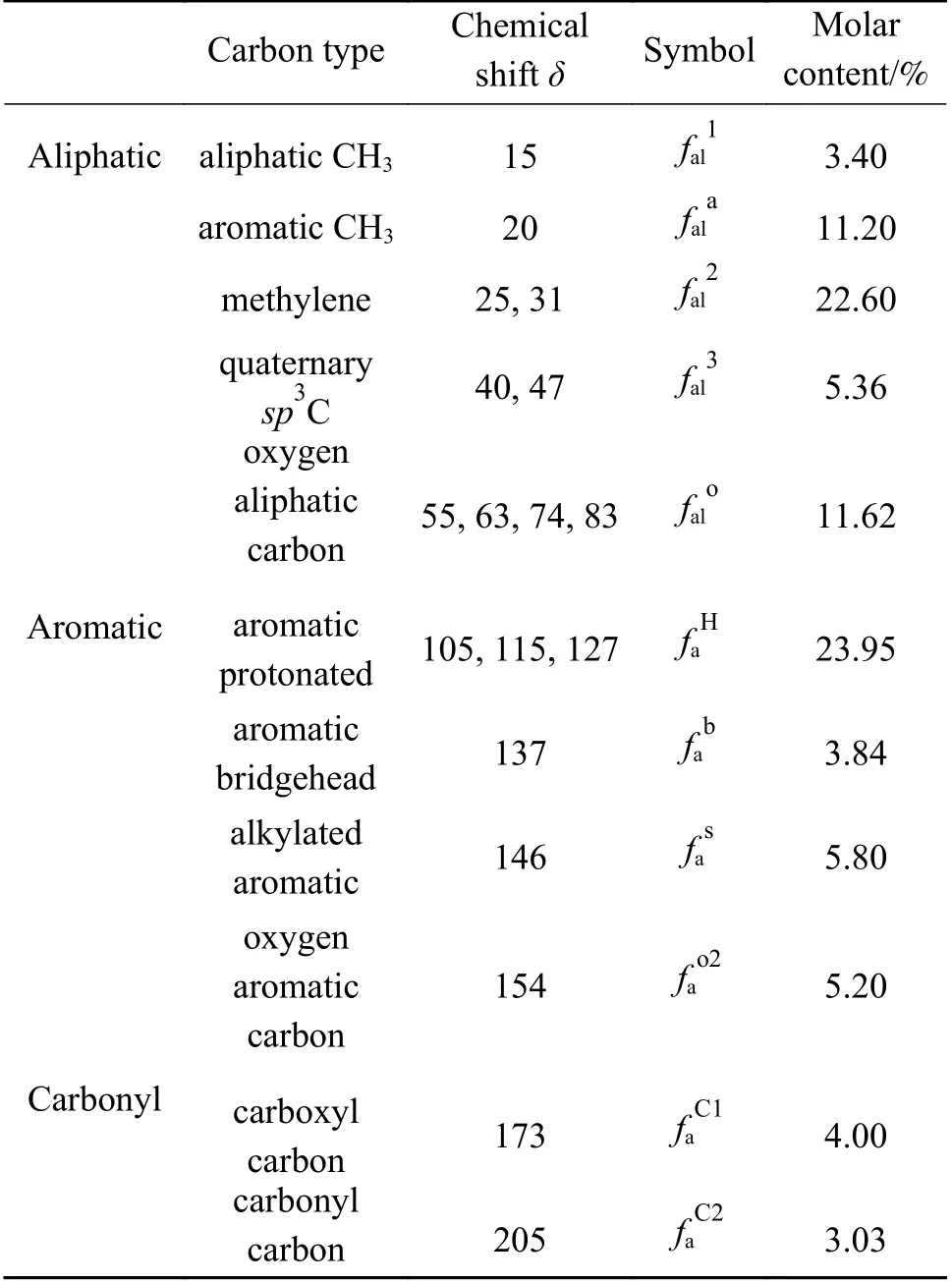
表2 ML 13C NMR分峰获得的含碳官能团的化学位移和含量Table 2 The chemical shifts and the contents of carboncontaining functional groups obtained from 13C NMR peaks of ML
2.2 傅里叶变换红外光谱分析
图3为ML的FT-IR谱图。其中,峰位的判断参考了相关文献[22-26]。在3413 cm-1处存在宽而强的吸收峰,其为-OH的伸缩振动;1710 cm-1处的小肩峰,来自于醛、酮、脂和羧酸的振动,这对应于13C NMR谱图中170-220处(羰基碳/羧基碳)的弱共振信号;在1603和1511 cm-1为芳烃特征吸收峰,且吸收峰明显,表明ML分子中富含芳香结构;1385、2920和2851 cm-1处为烷烃结构的C-H伸缩振动吸收峰,煤样在该部分的吸光度较强,说明ML含有大量的脂肪碳结构,这与13C NMR分析结果中,ML高脂肪族碳含量相一致;1030 cm-1处为脂肪醚/醇的吸收峰;1265 cm-1处为苯酚伸缩振动。在本研究中,FT-IR分析主要用于辨别官能团的种类,并与ML结构模型的计算谱图相比较,用于验证分子结构模型的准确性。
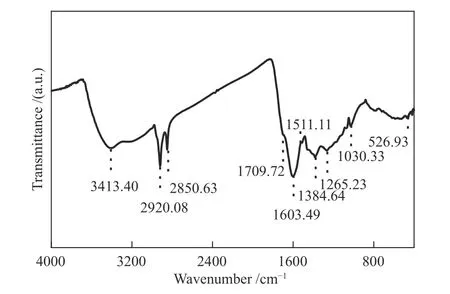
图3 ML的红外光谱谱图Figure 3 FT-IR spectrum of ML
2.3 X射线光电子能谱分析
XPS常用于分析煤中元素组成。参考相关文献[27-30],煤中的C主要存在于C-H/C-C、C-O、C=O/O-C-O以及COO-基团中;O主要存在于无机氧、C=O、C-O和COO-中;N主要以吡啶氮和吡咯氮的形式赋存。C 1s、O 1s和N 1s分峰拟合如图4所示,分析结果如表3所示。其中,峰位和结合能参考了相关文献[19,31-34]。
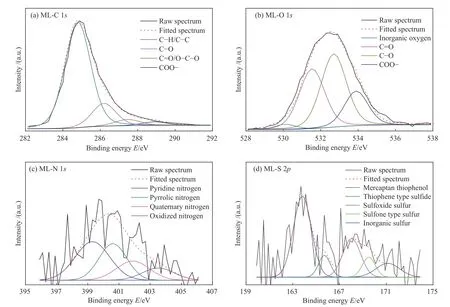
图4 ML的XPS分峰拟合图Figure 4 XPS peak fitting diagrams of ML
由图4与表3可知,ML的C-C/C-H含量高达77.22%,说明ML中的C主要以C-C/C-H的形式存在,这与经典的褐煤分子结构理论一致[10]。C 1s和O 1s分峰结果中均包含C-O、C=O/O-C-O和COOH结构。在C 1s中,C-O含量与C=O/O-C-O含量之比为3.46,而O 1s中C-O与C=O/O-C-O含量之比却仅为1.23。这归因于C=O/O-C-O中部分基团O-C-O的一个C连接了两个O,使得C=O/O-C-O在O 1s中响应强度较高,从而促使O 1s结果与C 1s不同。基于此,本研究结合了C 1s和O 1s的分析结果,在O 1s中除去无机氧后,得到剩余的C-O、C=O/O-C-O和COOH的总含量。按照C 1s中各含O组分的含量进行分配,最终得出无机氧、C-O、C=O/O-C-O和COOH的相对含量分别为2.45%、66.85%、19.31%和11.39%。从表3中N 1s和S 2p分析结果可以看出,N主要存在于吡啶氮(39.45%)和吡咯氮(30.22%)中,且与C、O的含量相比,N的含量较小,基于构建平均分子结构模型的理念,只考虑吡啶氮和吡咯氮。ML煤分子中有机硫主要存在于硫酚/硫醇中(48.82%),其次是亚砜型硫。本部分得出的各元素存在形式及其相对含量,也是构建ML分子结构模型的重要参数。
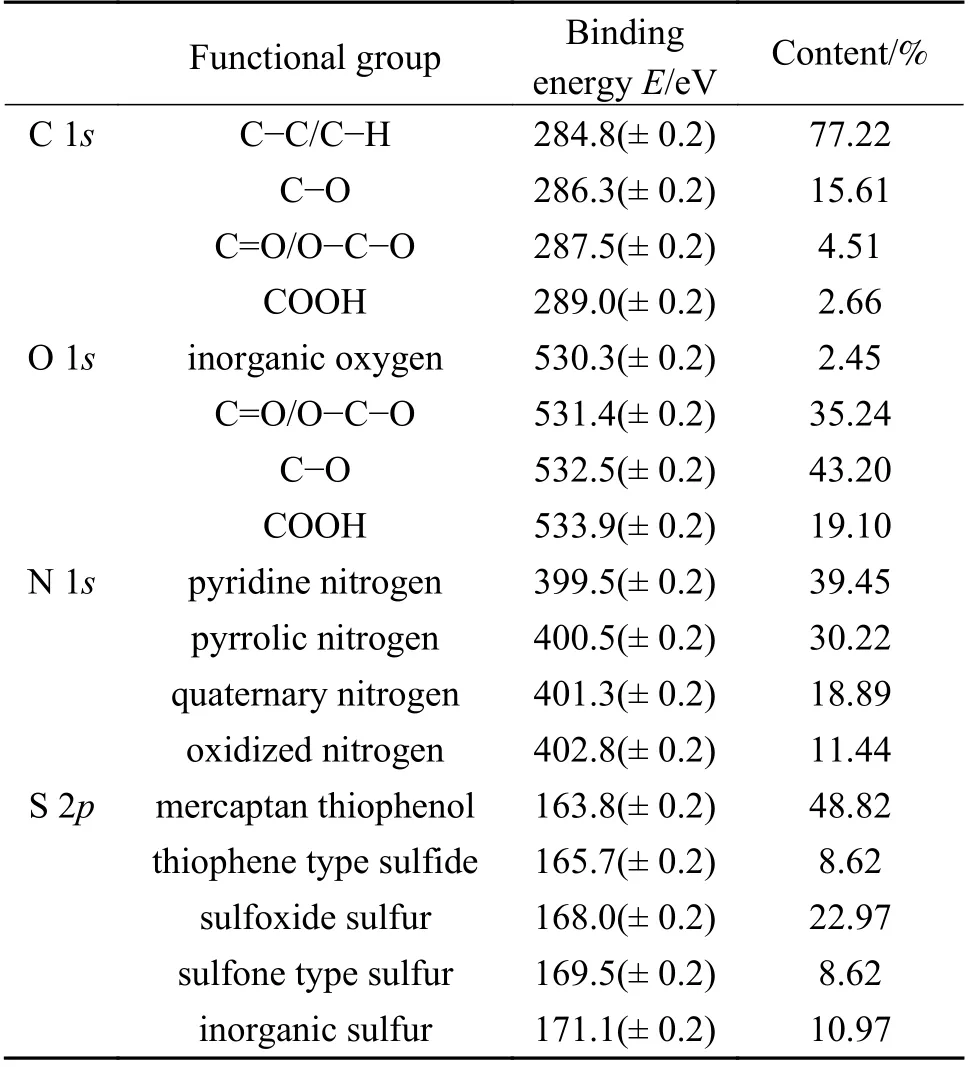
表3 ML的XPS分析Table 3 XPS analysis results of ML
2.4 ML的结构特性分析
目前,煤中碳原子数的确定方法主要是基于大分子结构单元的分子量,一般认为低阶煤分子的原子数目在200至400个,分子量为2500-3000[13,35]。此外,运行Gaussian 09程序的速度有限,过大的分子量会大幅延长计算时间。基于上述因素,本研究假定分子量为2500-3000,该分子量已经足够大,足以体现煤分子结构的复杂性和多样性[36]。
2.4.1 芳香碳结构
芳香碳结构的种类和数量通过芳香桥头碳与芳香周边碳的比值χb来确定,该参数反映了煤分子中芳香环缩聚程度的平均值。13C NMR分析结果表明,ML的χb为0.11。根据计算,苯和萘的χb分别为0、0.25。因此,苯和萘可以作为建模中的主要芳香族化合物。表3中N 1s分析表明,ML分子结构中N主要存在于吡啶和吡咯中,因此,在建模时须考虑吡啶和吡咯。通过调整苯、萘、吡啶和吡咯的数目,使得χb的值接近于0.11,最终得出的芳香结构的种类和数量如表4所示。其中,芳香碳原子为57个。根据13C NMR分析结果中ML的芳香度为38.79%,可以得出碳原子总量为147个。从元素分析结果表1中可得,H/C、O/C、N/C和S/C的原子数量比分别为1.0043、0.2431、0.0135和0.0045。因此,可以计算出H、O、N和S的原子数量分别为148、36、2和1。基于上述参数,可以确定ML结构模型的分子式为C147H148O36N2S。

表4 ML分子结构中芳香结构的种类和数量Table 4 Types and quantities of aromatic structures in ML molecules
2.4.2 脂肪碳结构
脂肪碳结构主要为环烷烃、烷基侧链以及氢化芳烃,煤阶越低,侧链越长[37]。根据碳原子总数以及13C NMR分析结果中脂肪碳所占比例,可以计算出甲基、亚甲基/次甲基、季碳和氧接脂碳的数量分别为21、33、8和17个。在构建模型的过程中,为了确保H/C比与元素分析结果一致,对脂肪碳原子的类型进行了适当的调整[11,12]。
2.4.3 杂原子结构
氧是褐煤中含量最丰富的杂原子,氮和硫是在褐煤的燃烧、热解以及转化利用等方面需重视的有害元素[37]。根据结构模型分子式可知,ML分子结构模型中氧原子数为36个,结合13C NMR、XPS(C 1s和O 1s)分析结果可得,羧基、羰基、醚氧以及酚羟基含量分别为6、5、17和2个。根据XPS中N 1s和S 2p的分析结果可得:氮原子主要存在于吡咯和吡啶中,两者均属于芳香结构,数量列于表4中;硫酚/硫醇含量占硫总量的48.82%,明显超其他形式有机硫的含量。由于ML模型分子式中硫原子仅有一个,在构建ML分子模型时,基于构建平均近似模型的理念,本研究选择硫醇代表煤分子中硫的平均类型。
2.5 ML分子结构模型的构建、优化与光谱模拟
2.5.1 ML模型的构建与优化
根据13C NMR、XPS以及元素分析等结果,确定了ML大分子碳骨架及侧链基团的种类和数量,将大分子碳骨架与侧链基团连接起来,即可构建出初始结构模型。由于初始构型局部能量较高且热力学稳定性较差,与平衡状态稳定的构型还有着较大的差异。因此,须不断地进行结构调整与构型优化,直至局部能量达到最小。本研究借助于Gaussian 09软件,采用半经验法MP3进行初步优化,然后采用密度泛函理论框架下的全电子M06-2X泛函对分子结构模型进行进一步优化,最终得出优化后的结构模型如图5所示。
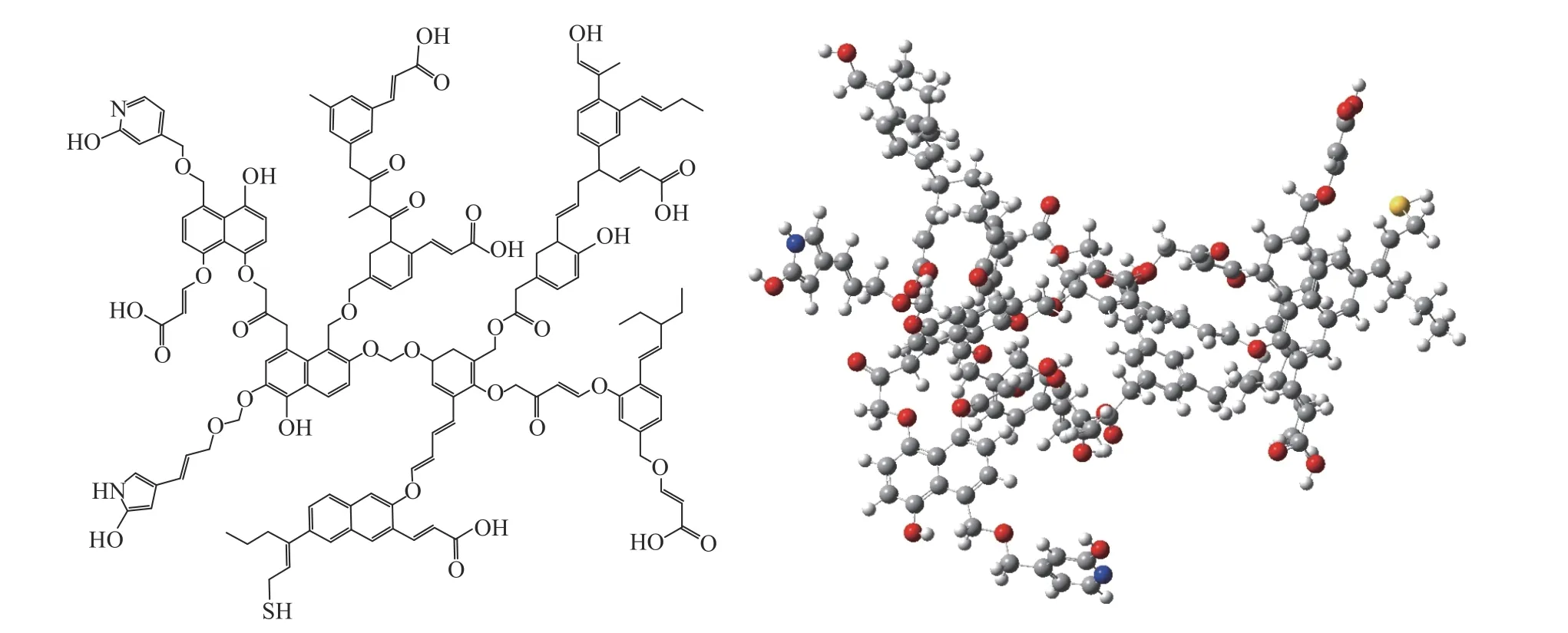
图5 ML的分子结构模型(C147H148O36N2S)Figure 5 Molecular structure model of ML (C147H148O36N2S)
经过构型优化,ML分子模型键长、键角发生变化,芳香碳骨架在空间中的排列也有较大变化。与中、高阶煤[6,9,13,38-40]相比,可以发现,在分子特征上,ML的大分子芳香碳骨架的环数较小,仅由单环的苯和两个环的萘构成,含氧官能团总量明显多于中、高阶煤;脂肪族侧链较多且较长,芳香度较低且结构有序度低;在空间构型上,ML的立体构型显著;芳香层片呈现空间上的不规则排列;杂原子官能团主要位于分子边缘。此外,由于ML结构中具有羧基氧,所以其可作为制取腐植酸的原料。并且ML含有大量桥键,在热解和加氢液化等过程中,桥键容易断裂,使其更容易转化为其他化工制品,有利于ML的综合开发利用。
2.5.2 ML结构模型的分子振动及核磁共振模拟
FT-IR光谱由分子振动和转动能级跃迁引起,因此,可将其简化为分子简谐振动。其中,计算方式主要包括密度泛函理论、从头算法以及半经验法。值得注意的是,由于模拟FT-IR是基于分子简谐振动近似计算的,需要用频率校正因子来修正谐振近似和理论方法的系统性误差。经过频率校正因子修正后,采用小基组、中等基组、大基组计算的结果几乎没有差别,但使用小基组却可以节省大量计算资源和时间。基于此,本研究选用小基组HF/3-21G来计算FT-IR谱图。图6(a)为ML分子模型的计算FT-IR谱图与实验FT-IR谱图的对比。由图6(a)可明显看出,计算出的FT-IR光谱与实验FT-IR光谱特征峰相似,总体吻合度良好,仅有小部分峰位和强度存在差异。其中,1000-1700 cm-1处的差异归因于含氧官能团伸缩振动引起的偏移[39]。实验FTIR谱图在3000-3600 cm-1处为宽峰,而计算FTIR谱图在此处为多个尖峰,这是由于计算的是单个分子,对于分子之间缔合氢键的伸缩振动无法计算到,所以在此区域以多个尖峰的形式呈现[40]。
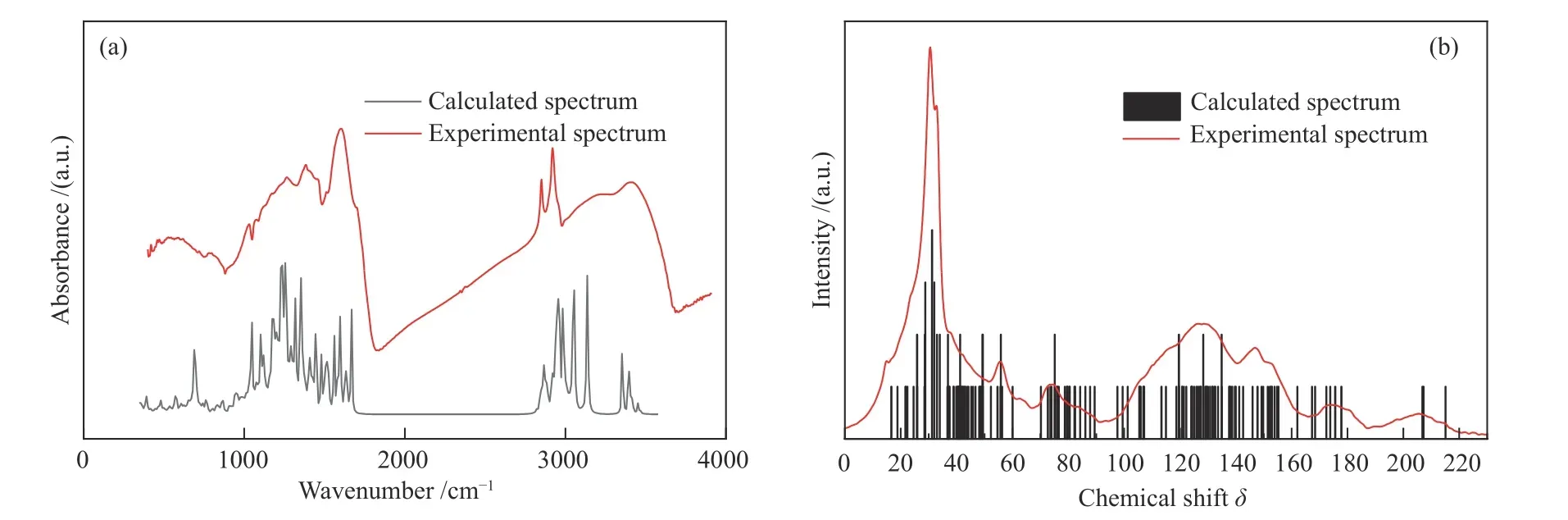
图6 计算谱图与实验谱图对比:(a)FT-IR谱图;(b) 13C NMR谱图Figure 6 Comparison of the calculated spectra with the experimental spectra: (a) FT-IR spectra; (b) 13C NMR spectra
使用密度泛函理论方法下的GIAO/6-31G(d, p)基组模拟计算获得的13C NMR波谱如图6(b)所示。不难看出,计算13C NMR谱图与实验13C NMR谱图吻合度较好。其中,化学位移δ为0-90处为脂肪碳部分;90-170 处为芳香碳部分;170-180 处为羧基碳;200-220 处为羰基碳部分。
计算FT-IR谱图和计算13C NMR谱图与实验谱图的良好吻合度证明了ML分子结构模型的准确性和合理性。
3 结 论
ML的芳香度fa为38.79%,芳香桥头碳/芳香周边碳χb为0.11,苯和萘是煤分子中主要的芳香族化合物。
根据元素分析及13C NMR分析,得出ML分子结构模型的分子式为C147H148O36N2S。
根据13C NMR和XPS分析结果,构建出ML的初始分子结构模型,借助于半经验法PM3和密度泛函M06-2X对初始模型进行构型优化进而得出了准确的分子结构模型。计算FT-IR光谱、计算13C NMR波谱分别与实验谱图相对比,吻合度良好,证明了ML分子模型的准确性和合理性。
ML分子结构中:氧主要以醚氧、羧基和羰基基团的形式存在;氮主要存在于吡咯和吡啶中;硫主要存在于硫醇/硫酚中。ML分子模型三维立体效果显著,侧链基团较多,且芳香层片在空间上呈现不规则排列,各芳香环之间主要通过甲基、亚甲基、甲氧基以及脂肪环的方式连接。
