Zr- Sn- Nb锆合金氧化膜中典型相结构的第一性原理研究
2021-07-28徐晨皓吴江桅谢耀平
赵 毅 王 栋 徐晨皓 吴江桅 王 洋 谢耀平
(1. 中国核动力研究设计院 反应堆燃料及材料重点实验室,四川 成都 610213; 2. 上海大学材料研究所,上海 200072;3. 上海大学计算机学院高性能计算中心,上海 200444)
锆合金是重要的核燃料包壳材料,耐腐蚀性是其关键性能指标,而锆合金氧化膜的性质决定着锆合金耐腐蚀性的好坏,因此研究锆合金氧化膜的性质具有重要意义。许多商用锆合金属于Zr- Sn- Nb系。Zr- Sn- Nb系合金氧化膜主要由单斜二氧化锆(monoclinic ZrO2, m- ZrO2)组成,还存在少量四方二氧化锆(tetragonal ZrO2, t- ZrO2)以及Sn和Nb的氧化物,如SnO、SnO2、NbO2和Nb2O5等。Preuss等[1- 2]的研究表明:锆合金氧化膜中不同位置t- ZrO2含量不同,距离金属/氧化膜(oxide/metal, O/M)界面越远,t- ZrO2含量越少;还通过电子背散射衍射研究了2种不同Sn含量锆合金(ZIRLO和Zr- 1.0Nb- 0.1Fe)氧化膜的相组成,发现氧化膜大部分物相为m- ZrO2,ZIRLO合金氧化膜中t- ZrO2含量比Zr- 1.0Nb- 0.1Fe合金的多。Wei等[3]研究了Sn含量对锆合金氧化膜相组成的影响,发现,在腐蚀过程中高Sn含量合金O/M界面形成了较多的t- ZrO2,随着腐蚀过程的进行,ZrO2向外扩展,形成了较多的m- ZrO2,而低Sn含量合金形成了较少的t- ZrO2。Frankel等[4]研究发现,在腐蚀过程中,ZIRLO合金、A- 0.6Sn合金以及A- 0.0Sn合金氧化膜中t- ZrO2的质量分数分别为4%~8%,4%~7%,2%~5%。
锆合金的耐腐蚀性与Sn含量密切相关,Sn含量降低,合金的耐腐蚀性提高[2]。Sn在锆合金中一般不以第二相的形式析出,通常偏聚在合金表面,不同初始取向锆合金表面Sn的偏聚程度也不同。Sn在氧化膜中有单质Sn、SnO2、SnO等存在形式[5- 6]。Takeda等[7]通过透射电子显微镜观察含Sn锆合金的氧化膜,发现m- ZrO2附近晶界存在SnO。
Nb在锆合金氧化膜中主要有2种存在形式:一是固溶于α- Zr或β- Zr;二是以第二相β- Nb存在。β- Nb是体心立方(body- centered cubic,BCC)结构,其氧化速率小于基体α- Zr[8]。β- Nb会氧化为非晶或四方相的二氧化铌(tetragonal NbO2, t- NbO2)[9- 10],或氧化为单斜相的五氧化二铌(monoclinic Nb2O5, m- Nb2O5)[11]。添加质量分数为0.1%的Fe的锆合金氧化膜中会出现Laves结构的第二相Zr(Nb,Fe)2,在距O/M界面0.8 μm处Laves相开始氧化为非晶态,距O/M界面1.5 μm处β- Nb开始氧化,这表明β- Nb具有更好的抗氧化性;Nb在靠近O/M界面为+2价,而靠近氧化物/水界面为+5价[12]。
目前有关Zr- Sn- Nb合金氧化膜性质的研究报道很多,但对氧化膜中Zr、Sn、Nb氧化物的形成与演化机制尚未完全解释清楚。因此,本文首先基于第一性原理计算了几种氧化物的形成焓,研究不同O化学势下氧化物的相对稳定性;然后结合热力学计算获得不同温度和压力下O化学势,得到氧化物的稳定性与压力和温度之间的关系;最后系统地确定了Zr、Sn、Nb氧化物形成的热力学驱动力随温度和压力的变化关系。
1 计算方法与模型构建
1.1 第一性原理计算方法
利用基于密度泛函理论(density functional theory, DFT)的第一性原理计算方法(first- principles method),对Zr、Sn、Nb氧化物的相结构总能进行计算,所有计算均采用vienna ab- initio simulation package(VASP)软件包实现[13]。采用投影缀加波(projector- augmented wave, PAW)描述[14- 15]离子与价电子之间的相互作用,采用广义梯度近似(generalized gradient approximation, GGA)下的Perdew- Burke- Ernzerhof(PBE)泛函处理电子间交换关联。k点网络采用Monkhorst- Pack方法产生[16]。计算过程中,对超胞的矢量和所有原子都进行弛豫,能量收敛标准为0.1 eV/nm。
1.2 参数测试
对平面波展开的能量截断进行计算。对于单质Zr,以密排六方(hexagonal close-packed, HCP)- Zr为对象,计算体系总能,其能量截断取100~450 eV,计算结果如图1(a)所示。当能量截断大于150 eV时,继续增加能量截断使总能的变化小于0.01 eV/atom。进一步测试能量截断对Zr- O体系形成焓的影响。根据O2、HCP- Zr和ZrO2在不同能量截断下的总能,计算m- ZrO2的形成焓,结果列于图1(b)。由图1(b)可以看出,当能量截断达到500 eV时,继续增加能量截断,形成焓的变化小于0.01 eV/atom。
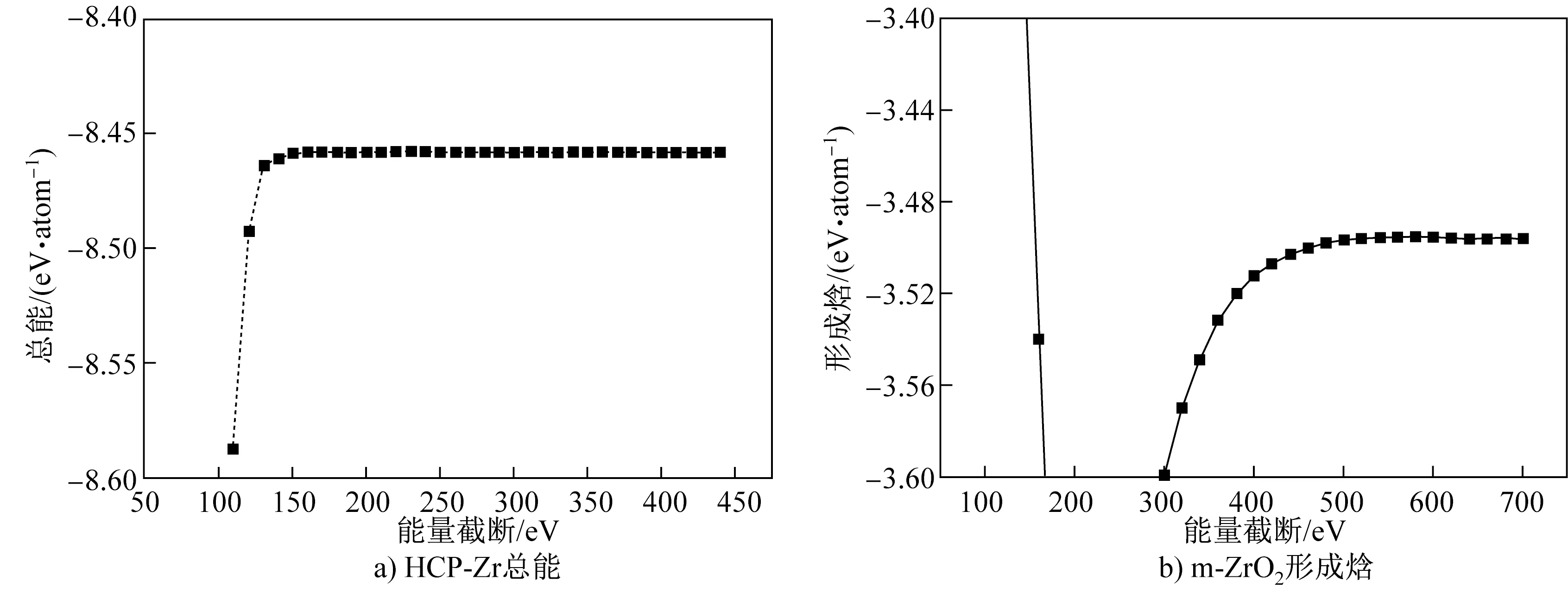
图1 HCP- Zr总能与m- ZrO2形成焓随能量截断的变化
采用Monhkorst- Packk点取样方法对布里渊区进行积分,根据HCP- Zr(a=0.323 nm,c=0.515 nm)晶胞形状,分别取(2×2×1)、(4×4×2)、(6×6×4)、(8×8×6)、(10×10×6)、(12×12×8)、(14×14×8)等k网格点,测得不同k点网格密度下的总能如图2所示。由图2可以看出,当k点网格密度达到(6×6×4)时,继续增加网格密度,总能变化小于0.01 eV/atom。因此,参照HCP- Zr体系的结果,本文所有k点取样均按照密度相等的原则进行设置。
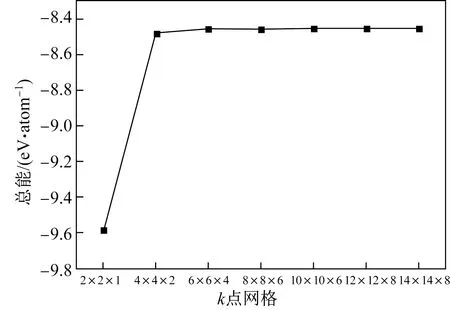
图2 HCP- Zr总能随k点网格密度的变化
2 计算结果与讨论
2.1 典型氧化物的晶体结构与形成焓
Zr- Sn- Nb系合金氧化膜中典型氧化物有m- ZrO2、t- ZrO2、t- SnO、t- SnO2、NbO2和m- Nb2O5,氧化物原胞如图3所示。计算选取截断能为500 eV,k点网格分别为7×7×7、9×9×7、2×2×1、1×1×1、1×1×2、1×1×1。通过晶格优化获得的氧化物晶格常数如表1所示,可知m- ZrO2和t- ZrO2的晶格常数计算值与试验值吻合较好。Sn和Nb的氧化物的晶格常数与m- ZrO2相差较大,因此氧化物界面为非共格关系,界面能对微结构的影响较大。形成焓(Hf)可用来反映氧化物的稳定性,其计算公式[17]为:

表1 Zr- Sn- Nb系合金氧化膜中氧化物的晶格常数a, b, c及晶胞矢量夹角α, β, γ

图3 Zr- Sn- Nb系合金氧化膜中氧化物原胞(红色小球代表O原子,其余小球代表金属原子)
(1)
式中:Etot为晶胞总能;EM(M=Zr,Nb,Sn)和EO分别为金属元素M和O基态时的平均单个原子能量,基态下Zr为HCP结构,Nb为BCC结构,Sn为面心立方(face- centered cubic, FCC)结构,O为氧分子结构;ntot为晶胞中原子总数;nM和nO分别为晶胞中M、O原子的个数。
根据式(1),通过第一性原理计算得到晶胞的总能以及M、O元素在其平衡态晶体结构中的平均单个原子能量,结果列于表2。由表2可知,m- ZrO2的形成焓最低,SnO的形成焓最高,同种金属不同类型氧化物的形成焓差值较小,均小于0.21 eV/atom。不同金属氧化物的形成焓差值较大,Zr氧化物的形成焓比Nb氧化物的约低1.38 eV/atom,而Nb氧化物的形成焓比Sn氧化物的约低1.1 eV/atom。根据形成焓的计算结果可大致判断出:Zr的氧化驱动力最大,Nb的次之,Sn最难氧化,其中Nb的氧化慢于Zr,与试验结果相吻合[18]。在NIST- JANAF数据中NbO2和Nb2O5的形成焓分别为-2.73和-2.79 eV/atom,比计算值约大0.5 eV/atom,表明DFT对NbO2和Nb2O5中Nb—O化学键有所低估,这与著名O—O化学键低估问题类似[19]。

表2 由第一性原理计算得到的Zr- Sn- Nb合金氧化膜中氧化物的形成焓
2.2 氧化物稳定性与O化学势的关系
金属氧化物的形成焓是反映其稳定性的关键物理量,与从环境中获取O原子的难易程度有关(即O化学势),随O化学势的变化而变化。氧化物形成焓的计算公式为:
(2)
式中μO为氧的化学势。
图4为氧化膜中Zr、Sn和Nb氧化物的形成焓随O化学势的变化。可以看出,O化学势越高,金属越容易从环境得到氧。比较发现,价态相同的氧化物,如m- ZrO2、t- ZrO2、t- SnO2、t- NbO2的形成焓差值不随化学势变化而变化,而价态不同的氧化物形成焓的差值则随化学势变化而变化。

图4 Zr- Sn- Nb合金氧化膜中Zr、Sn和Nb氧化物的形成焓随O化学势的变化
由于O化学势变化不会引起m- ZrO2、t- ZrO2、t- SnO2、t- NbO2形成焓的变化,因此也不会引起Zr、Nb和Sn氧化顺序的变化。由此可以推测,试验t- ZrO2相成分的改变不是O化学势变化引起的,而是其他原因,如压力、晶粒大小等引起的。O化学势变化会引起NbO2和Nb2O5的相对稳定性改变,富氧区和贫氧区对应的O/M界面侧和O/W水侧的产物也有所变化,O/M界面附近稳定相是NbO2,而O/W附近则为Nb2O5,这与碱性水中氧化膜的试验结果相吻合[18],也与氧化性相对弱的去离子水中出现NbO2,而氧化相对较强的碱性水常出现Nb2O5的结论相一致[20]。进一步推测出,O/M界面附近Sn氧化物稳定相是SnO,而O/W附近则为SnO2。因此,根据上述结果可以定性地分析氧化膜不同位置的相种类。
2.3 温度与压力对O化学势和氧化物相对稳定性的影响
通过热力学公式计算特定温度和压力下的O化学势,得到一定温度和压强下氧化物的相对稳定性,计算公式为:
(3)
(4)

对于腐蚀试验,常用的2个水化学条件为360 °C/18.6 MPa去离子水以及400 ℃/10.3 MPa过热水蒸气。计算得到10.3和18.6 MPa压力,300~500 ℃(573~773 K)温度下O化学势分别为-7.36~-7.22和-7.38~-7.14 eV/atom。图5为在10.3 MPa和300~500 ℃(573~773 K)条件下,O化学势随温度的变化以及氧化物形成焓随O化学势的变化。可以看出,NbO2和Nb2O5在O化学势为-7.01 eV/atom时的形成焓相等,在低于-7.01 eV/atom的贫氧侧,NbO2更稳定,而在高于-7.01 eV/atom的富氧侧,Nb2O5更稳定,这与O越多,Nb2O5越容易生成是一致的。O化学势为-7.01 eV/atom时,对应10.3 MPa、500 ℃以上的化学势。因此,从热力学角度分析,在10.3 MPa、300~500 ℃(673~773 K),Nb的氧化物稳定相是NbO2,这与部分试验得到的去离子水中Nb氧化物为NbO2一致[20],但与另一部分试验得到的Nb氧化物为Nb2O5不一致[18],这可能是试验条件复杂,部分条件没有在计算中体现所致。SnO和SnO2在O化学势为-7.12 eV/atom时的形成焓相等,在低于-7.12 eV/atom的贫氧侧,SnO更稳定,在高于-7.12 eV/atom的富氧侧,SnO2更稳定。O化学势为-7.12 eV/atom时,对应10.3 MPa、500 ℃以上的化学势。因此,在10.3 MPa、300~500 ℃(573~773 K),Sn的氧化物稳定相是SnO,这能解释Takeda等[7]的试验结果;在18.6 MPa、300~500 ℃(573~773 K),Nb和Sn氧化物的稳定相仍分别是NbO2和SnO。

图5 10.3 MPa压力下O化学势随温度的变化以及氧化物形成焓随O化学势的变化
图6为在327 ℃(600 K)和10~20 MPa条件下,O化学势随压力的变化。由图6可知,600 K下,10~20 MPa压力对应的O化学势为-7.262~-7.244 eV/atom。O化学势随压力的增加而升高,压力越大,金属越容易从系统中获得O原子,因此压力越大,锆合金越容易被氧化。NbO2和Nb2O5在O化学势为-7.01 eV/atom时的形成焓相等,对应的压力大于20 MPa;SnO和SnO2在O化学势为-7.12 eV/atom时的形成焓相等,对应的压力也大于20 MPa。在600 K和10~20 MPa压力范围内,SnO比SnO2稳定,NbO2比Nb2O5稳定。因此,在此压力范围内稳定氧化物的种类不随压力而改变,氧化膜中Nb和Sn的氧化物稳定相仍是NbO2和SnO。

图6 600 K下O化学势随压力的变化
3 结论
(1)Zr- Sn- Nb合金氧化膜中,m- ZrO2的形成焓最小,最稳定,这与试验得到的氧化膜主要物相为m- ZrO2相一致。氧化物稳定性由强到弱的顺序为Zr氧化物、Nb氧化物和Sn氧化物,氧化膜中典型析出相β- Nb的氧化速率小于Zr基体,与试验结果相吻合。
(2)Nb和Sn氧化物的稳定性随O化学势的变化而变化,贫氧区NbO2和SnO比较稳定,富氧区Nb2O5和SnO2比较稳定,这与试验中NbO2常出现于氧化性较弱的去离子水环境中,而Nb2O5常出现于氧化性较强的碱性水中的结论相一致。
(3)在10.3和18.6 MPa压力、300~500 °C(573~773 K)温度条件下,Sn的稳定相均为SnO,Nb的稳定相均为NbO2,能解释一些试验结果。
