拉曼Mapping成像研究聚苯乙烯/聚甲基丙烯酸甲酯共混体系的相态结构
2021-07-28陈韶云田杜李奇钟敏胡成龙纪红兵
陈韶云,田杜,李奇,钟敏,胡成龙,纪红兵
(1 江汉大学化学与环境工程学院,光电化学材料与器件教育部重点实验室,湖北武汉 430056;2 中山大学化学学院精细化工研究院,广东广州 510275)
高分子材料科学发展至今,获得高分子多相体系材料的常用方法是聚合物共混,即通过物理或化学方法将两种或多种聚合物共混形成一种具有新性能材料的方法。这种方法的优点在于综合利用了两种或多种聚合物材料固有的属性,从而提高材料的力学性能或光学性能等。事实上,绝大多数聚合物是不相容或部分相容的,属热力学不相容体系。该体系存在两个问题,一是组分之间的相容性差,界面黏合性也随之变差,两相不稳定,容易出现相分离;二是分散相在连续相中的尺寸大小不均一、分布不均匀,导致共混体系的内部结构不均匀。因此,聚合物的非均相体系在微观尺度上的化学组成和形态结构在很大程度上决定了共混材料的性能[1-3]。改善相容性获得结构稳定以及性能优异的聚合物共混材料主要采用增容技术:一是在共混体系中加入非反应性嵌段或接枝共聚物作为增容剂(物理增容)[4-5];二是在共混体系中引入其他化学分子与共混组分发生化学反应来提高共混体系的相容性(反应性增容)[6-7]。在增容加工过程中微观纳米尺度下体系的组分和相态结构随着加工条件的改变或化学反应的进行不断演化,而研究聚合物共混体系的相态结构或相界面的演变有助于聚合物共混体系增容剂的选择及定量、界面反应动力学参数测定,从而确定相应的加工条件和反应增容条件。
聚合物共混体系的微观组分、结构、形态分析等的测试表征技术主要有X 射线光电子能谱(XPS)[8]、二次离子质谱(SIMS)[9]、中子散射谱(NSS)[10]、扫描探针显微镜(SPM)[11]、扫描电子显微镜(SEM)、透射电子显微镜(TEM)和原子力显微镜(AFM)等。尽管这些表征技术在相界面结构的研究中已被广泛应用,然而它们在提供详细的界面结构信息方面常常缺乏足够的表面专一性或灵敏性。如XPS是最灵敏的表界面技术之一,但仍会受到材料本体的干扰,本体的信号甚至会超过表界面产生的信号,而且主要应用在表界面化学成分分析上,且检测需要超高真空环境,也不能提供相界面的微观相态形貌与分布;SEM、TEM 和AFM可以表征复合材料体系的微观相态形貌与分布,但难以识别样品中单个粒子相的化学结构信息[12]。近几年来,分子光谱技术发展迅猛,这种技术本身具有超高灵敏性、高度选择性,不仅能实时地提供界面分子的结构信息、成分信息和分子基团取向信息,更重要的是通过分子光谱成像可以得到复合体系(或共混体系)的相界面的微观相态形貌与分布,如纳米红外技术是近年来兴起的一种有效的微区形貌及化学成分成像工具,其空间分辨率可以达到10nm[13],Ye 等[14]利用AFM-IR 技术在聚碳酸酯/丙烯腈苯乙烯/丁二烯(PC/ABS)共混体系中检测到40nm 直径的软聚丁二烯析晶。但是纳米红外技术一般要与AFM 联用,不仅成本高,而且在一般实验室也难以实现。
拉曼光谱技术(Raman spectroscopy,RS)作为一种非破坏性的分子光谱检测技术,不仅可在几到几十微米空间分辨率下提供非均相材料特有的分子指纹信息,而且其Mapping 成像技术采用直接原位成像技术来研究相界面并测量出相应的界面参数,其测量范围可以从微米量级扩展至几百纳米,迅速成为了研究物质表界面的一种有力工具[15-18]。Morgan 等[19]利用拉曼Mapping 成像技术研究了线性氘代聚乙烯/支化聚乙烯共混体系的形态结构,当共混体系淬火之后,Mapping 成像图可以反映出共混体系各组分的结晶变化;Huan 等[20]用共焦显微拉曼成像技术研究了聚对苯二甲酸乙二醇酯/高密度聚乙烯(PET/HDPE)共混体系及其增容体系的相态结构和化学组成分布,发现该不相容体系发生相分离导致化学结构组成呈非均一性,当体系加入增容剂顺丁烯二酸酐后,PET和HDPE之间的黏性和分散性明显提高;Xue 等[21]将高空间分辨针尖增强拉曼成像技术(TERS)应用于(苯乙烯-丙烯腈)(PMMA/SAN)共混薄膜的相结构分析,通过局域部位的化学相识别检测到PMMA/SAN 共混体系中的相分离行为,并直观地观察到早期相演变约200nm 的界面宽度。这些工作表明,拉曼光谱成像技术可以用来研究共混体系的化学结构和物理形貌等多种信息。然而拉曼成像技术在聚合物共混物中的应用并不广泛,主要原因是目前还未有系统的研究方法和实验结果及相应的理论模型。因此本文选用非晶的聚苯乙烯(polystyrene, PS)和聚甲基丙烯酸甲酯[poly(methyl methacrylate),PMMA]的共混体系作为研究对象,原因在于PS/PMMA 共混物是典型的热力学不相容性体系[22-26],当体系作为共混增容研究对象时,作为增容剂的嵌段共聚物或接枝共聚物(如嵌段聚合物PS-b-PMMA)容易制备,通过拉曼光谱成像技术来研究PS/PMMA 共混体系及其增容体系的相态结构与分布和性能之间的关系,形成系统研究相态结构的拉曼光谱成像方法,从而为新型聚合物共混合金的产品性能控制或应用提供一定的理论依据和实际指导。
1 实验
1.1 试剂与仪器
聚苯乙烯(PS,M=171000,Mw/Mn=1.9)、聚甲基丙烯酸甲酯(PMMA,M=57000,Mw/Mn=1.56)、苯乙烯(St)、甲基丙烯酸甲酯(MMA)、四氢呋喃(THF)、甲醇、二氯乙烷、氢氧化钠和无水硫酸钠均为分析纯,国药集团化学试剂有限公司;2-溴丙酸乙酯(纯度98%)、2,2-联吡啶(bpy,纯度99.0%)、氯化亚铜(CuCl,纯度≥99.95%),上海阿拉丁试剂公司。激光显微拉曼光谱仪,型号inVia英国雷尼绍公司。热重分析仪,型号TG 209 F3,德国Netzsch。差示扫描量热仪,型号Q-20,美国TA 公司。凝胶渗透色谱仪,型号PL-GPC50,Polymer Laboratories(PL)公司。
1.2 实验过程
1.2.1 嵌段共聚物PS-b-PMMA的制备
(1)大分子引发剂PS-Br的合成 采用原子转移自由基聚合(ATRP)的方法制备大分子引发剂PS-Br,即0.001mol CuCl、0.002mol bpy 和0.1mol苯乙烯单体加入到50mL的两口瓶中充分溶解,静置10min 后,向上述体系中加入0.001mol 2-溴丙酸乙酯后并通入氮气,室温搅拌1h,然后将体系的温度升至110℃,反应12h,反应方程如式(1)。最后体系自然冷却至室温,加入THF 超声溶解产物,离心沉淀除去无机盐,将上层清液用甲醇沉淀,得到纯净的PS-Br 大分子引发剂。所制备PSBr,Mn=6906,Mw=10886,Mw/Mn=1.58,玻璃化转变温度Tg=76℃。核磁氢谱:δ7.04(m,—C6H5),δ1.759(m,—CH),δ1.338(m,—CH2—)。氘代氯仿的溶剂峰出现在7.26处,7.04对应于苯环上的5个氢,1.759对应于次亚甲基上的氢,1.338是亚甲基上的氢。

(2)嵌段聚合物PS-b-PMMA的合成 采用已合成的PS-Br作为大分子引发剂(0.4g),2,2-联吡啶(0.048g)和氯化亚铜(0.015g)作为配合催化剂,甲基丙烯酸甲酯为反应单体,高纯氮气作为保护气,在90℃的条件下实现原子转移自由基聚合,反应方程如式(2)。最后体系自然冷却至室温,加入THF 超声溶解产物,离心沉淀除去无机盐,将上层清液用甲醇沉淀,得到纯净的PS-b-PMMA大分子引发剂。所制备的PS-b-PMMA,Mn=32609,Mw=96643,Mw/Mn=2.96,玻璃化转变温度Tg=79℃(PS 链段),Tg=114℃(PMMA 链段)。核磁氢谱:δ7.08 (m,—C6H5),δ3.599 (s,—OCH3),δ0.84、δ1.02(m,—CH3)。化学位移7.08对应PS苯环上五个氢原子的共振峰,3.599对应于PMMA中—OCH3的氢原子,0.84和1.02对应于PMMA中—CH3的氢原子,1.160~2.100 对应于聚合物主链上的氢原子的共振峰。PS-Br-PMMA 中PS 的质量分数为21.2%。

1.2.2 PS/PMMA共混薄膜及其增容体系的制备
(1)PS/PMMA 共混薄膜的制备 采用溶液共混的方法制备PS/PMMA 薄膜,即分别将25g PS 和PMMA溶于250mL二氯乙烷中,充分溶解之后形成0.1mg/mL 的PS 和PMMA 溶液。取5mL PS 溶液和5mL PMMA 溶液于试管中充分混合形成质量比为PS/PMMA=50/50的混合溶液,然后用移液管取3mL上述混合溶液滴涂在2cm×2cm 的载玻片上,使混合溶液充满整个载玻片,自然干燥成薄膜,薄膜厚度2~3μm。最后将所制成的薄膜置于120℃真空干燥箱中退火12h,使PS/PMMA 薄膜充分发生相分离。 用同样的方法制备PS/PMMA=30/70 和PS/PMMA=70/30薄膜样品,如图1所示。
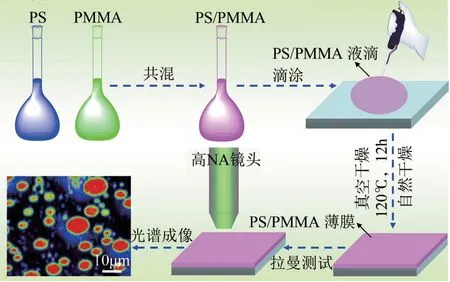
图1 PS/PMMA共混薄膜的制备过程及其拉曼光谱成像示意图
(2)PS/PMMA 增容体系的制备 将5g PS 和5g PMMA 溶于50mL的二氯乙烷中,充分溶解之后形成0.1mg/mL PS 和0.1mg/mL PMMA 的混合溶液,此时PS 和PMMA 的质量比为50:50,然后在上述混合溶液中加入0.5g PS-b-PMMA 作为体系的增容剂,充分溶解之后得到质量比为PS/PMMA/PS-b-PMMA=50/50/5 的混合溶液。用移液管取3mL 上述混合溶液滴涂在2cm×2cm 的载玻片上,使混合溶液充满整个载玻片,自然干燥成薄膜。最后将所制成的薄膜置于120℃真空干燥箱中退火12h,使PS/PMMA 薄膜充分发生相分离和消除薄膜内部应力。用同样的方法制备PS/PMMA/PS-b-PMMA=30/70/5 和 PS/PMMA/PS-b-PMMA=70/30/5 薄膜样品。
1.2.3 PS/PMMA 共混薄膜及其增容体系的拉曼成像测试
采用共焦显微拉曼光谱仪记录光谱信息和图像处理,激光器波长为532nm(半导体激光器),物镜为50倍,0.75N.A.Leica 物镜,光谱用硅片进行校正(校正散射频率为520cm-1),空间分辨率为2μm。光谱成像模式采用静态扫描模式,以1150cm-1为光谱扫描中心,波数范围为200~2000cm-1,曝光时间3s,累计次数1 次,扫描步径2.0μm,如图1所示。
2 结果与讨论
前人的研究表明PS 和PMMA 是热力学不相容体系[27-28],这是因为在聚合物共混体系中,聚合物分子链间的范德华力作用不利于组分之间相互溶解和渗透,其混合焓通常大于零,而混合熵不足以克服混合热形成均相体系[29]。另外由于PS 和PMMA分子之间不存在特殊的相互作用,如氢键、离子-离子或电荷转移络合等,因此PS 和PMMA 属于热力学不相容体系。图2(a1)~(c1)分别是PS/PMMA 质量比为30/70、50/50、70/30 共混薄膜的光学显微图,PS 和PMMA 发生了明显的相分离,形成典型的“海岛”结构,即一相为连续的“海相”,另一相为分散的“岛相”。本文中PS/PMMA共混薄膜采用溶液法制备,相比于螺杆加工而成的PS/PMMA共混体系,该体系没有受到剪切力作用,因此体系的相态结构在显微镜下能观察到,但是仍不能得到精确的化学结构和相分布信息。为了得到PS/PMMA共混薄膜化学成分的精确分布图,采用点到点扫描成像模式(point-to-point mapping)来记录每个扫描点的拉曼光谱,然后以各组分的特征拉曼散射峰的强度作图可以得到拉曼Mapping图。PS的特征频率为1001cm-1,PMMA 的特征频率为814cm-1[30],如图2(a2)~(c2)所示,共混体系的Mapping 图相比于光学显微镜成像,两相结构的图像更直观清晰,分散相为球形结构。此外,从图中可以看出PS 和PMMA 无论以何种比例混合,共混体系的相态结构呈现出明显的相分离现象。众所周知,相容性被用来描述分子水平的最大混合程度,对于聚合物大分子来说,聚合物的分子尺寸比较大,所以绝大多数聚合共混体系都达不到分子水平或链段水平互容,但是很多非均相体系仍然可以看作是互容体系,这是因为高分子共混体系的分散程度取决于组分间的相容性。聚合物的相容性或均一性与实验探针尺寸(experimental probe size)和相区尺寸(domain size of the phase)相关,相容性指数可以定义为式(3)[31]。

图2 聚合物共混体系的光学图、拉曼光谱成像图和粒径分布图

式中,相区尺寸是指共混体系中分散相的平均尺寸。当Nc为无穷大时(分散相很小或分散相不存在形成均一体系),共混体系为完全相容;当Nc=1,共混体系为半相容;当Nc=0,共混体系为完全不相容。当PS/PMMA=30/70,分散相尺寸范围为4.0~14.0μm,平均尺寸约为7.5μm,Nc=0.26;当PS/PMMA=50/50,分散相尺寸范围为3~11μm,分散相平均尺寸为6.0μm,Nc=0.33;当PS/PMMA=70/30,分散相尺寸范围为4.0~18.0μm,分散相平均尺寸为8.0μm,Nc=0.25。由计算结果可知,PS和PMMA 无论以何种比例混合,该共混体系都是热力学不相容体系,与拉曼Mapping的结果一致。

图3 聚合物共混体系的光学图、拉曼光谱成像图和粒径分布图
聚合物共混体系在应用时,存在组分间力传递和相态稳定性的问题,如果组分间的相容性不好,不但应力传递受阻,而且共混体系的相态结构也不稳定,因此提供组分间相容性是关键,其目的在于降探低界面张力,促进组分分散程度的提高,进而提高相态结构的稳定性,改善共混聚合物的力学性能。本文利用非反应性嵌段共聚物PS-b-PMMA 作为增容剂来改善PS 和PMMA 共混体系的相容性。图3(a1)、(b1)和(c1)是PS/PMMA 共混体系的光学显微图,无论组分以何种比例混合,加入增容剂后,分散相的尺寸变小而且均匀的分布在连续相中;图3(a2)、(b2)和(c2)是不同比例混合的PS/PMMA/PS-b-PMMA 增容共混体系的拉曼Mapping 图,与光学显微图相对应,分散相以“海岛状”的形式均匀地分布在连续相中,当PS/PMMA/PS-b-PMMA=30/70/5,分散相尺寸范围为3.0~6.0μm,平均尺寸约为4.5μm,Nc=0.44;当PS/PMMA/PS-b-PMMA=50/50/5,分散相尺寸范围为1.5~5.0μm,分散相平均尺寸为3.5μm,Nc=0.57;当PS/PMMA/PS-b-PMMA=70/30/5,分散相尺寸范围为3.0~6.0μm,分散相平均尺寸为4.0μm,Nc=0.5。由计算结果可知,相比PS/PMMA 共混体系,PS/PMMA/PS-b-PMMA=50/50/5共混体系的相容性指数提高了近2倍,说明PS-b-PMMA的加入能有效改善PS和PMMA的相容性。增容机理是嵌段共聚物PS-b-PMMA 可以看作是一个“刷子”,由于PS和PMMA是高度不相容体系,因此PS-b-PMMA趋向分布在PS和PMMA共混体系的界面[32],“刷子”中的PS 链段和组分PS 相容,PS-b-PMMA 中的PMMA 链段和组分PMMA 相容,起到连接桥梁的作用,大大降低了共混体系的界面张力[33]。
在聚合物共混体系中鉴别分散相和连续相对材料的设计制备具有重要的意义,传统的扫描电子显微镜只能观察分散相和连续相的微观结构,无法鉴别分散相和连续相的组分构成。拉曼Mapping成像技术既可以观察共混体系的相态结构,又可以测得分子光谱来鉴别组分的构成,在图2(a2)~(c2)中分别取分散相、界面相和连续相的拉曼光谱。如图4所示,1001cm-1是PS 分子链中苯环的呼吸振动(或伸缩振动),814cm-1是PMMA中C=O的伸缩振动。在PS/PMMA=30/70共混体系中[图2(a2)],分散相1处(红色区域)的拉曼光谱如图4(a)中的曲线1所示,分子光谱中PS 特征峰的散射强度远大于PMMA 特征峰的强度;界面相2 处(绿色界面区域)的拉曼光谱如图4(a)中的曲线2 所示,分子光谱中PS 特征峰的散射强度相当于PMMA 特征峰的强度;连续相3处(深紫色区域)的拉曼光谱如图4(a)中的曲线3所示,分子光谱中PS特征峰的散射强度远弱于PMMA 特征峰的强度,说明该体系中PS 是分散相,PMMA 是连续相。在PS/PMM=50/50共混体系中[图2(b2)],分散相1 处(红色区域)的拉曼光谱如图4(b)中的曲线1所示,分子光谱中PS特征峰的散射强度远大于PMMA 特征峰的强度,说明该球状离子为PS;界面相2 处(绿色界面区域)的拉曼光谱如图4(b)中的曲线2 所示,分子光谱中PS 特征峰的散射强度相当于PMMA 特征峰的强度,说明该界面区是两相共存区;连续相3处的拉曼光谱如图4(b)中的曲线3所示,分子光谱中PS特征峰的散射强度相当于PMMA 特征峰的强度,说明分散相中既含有PS 也含有PMMA,可能是由于共混体系中PS 和PMMA 的质量分数相同。由于PS 是刚性链,一部分PS 分子链聚集成球状分散在共混体系中,另一部PS 分子链仍然存在于PMMA连续相中。在PS/PMMA=70/30 共混体系中[图2(c2)],由于组分的比例发生翻转,共混体系的微观相态结构也发生翻转,如图4(c)所示,分散相是PMMA,连续相是PS。
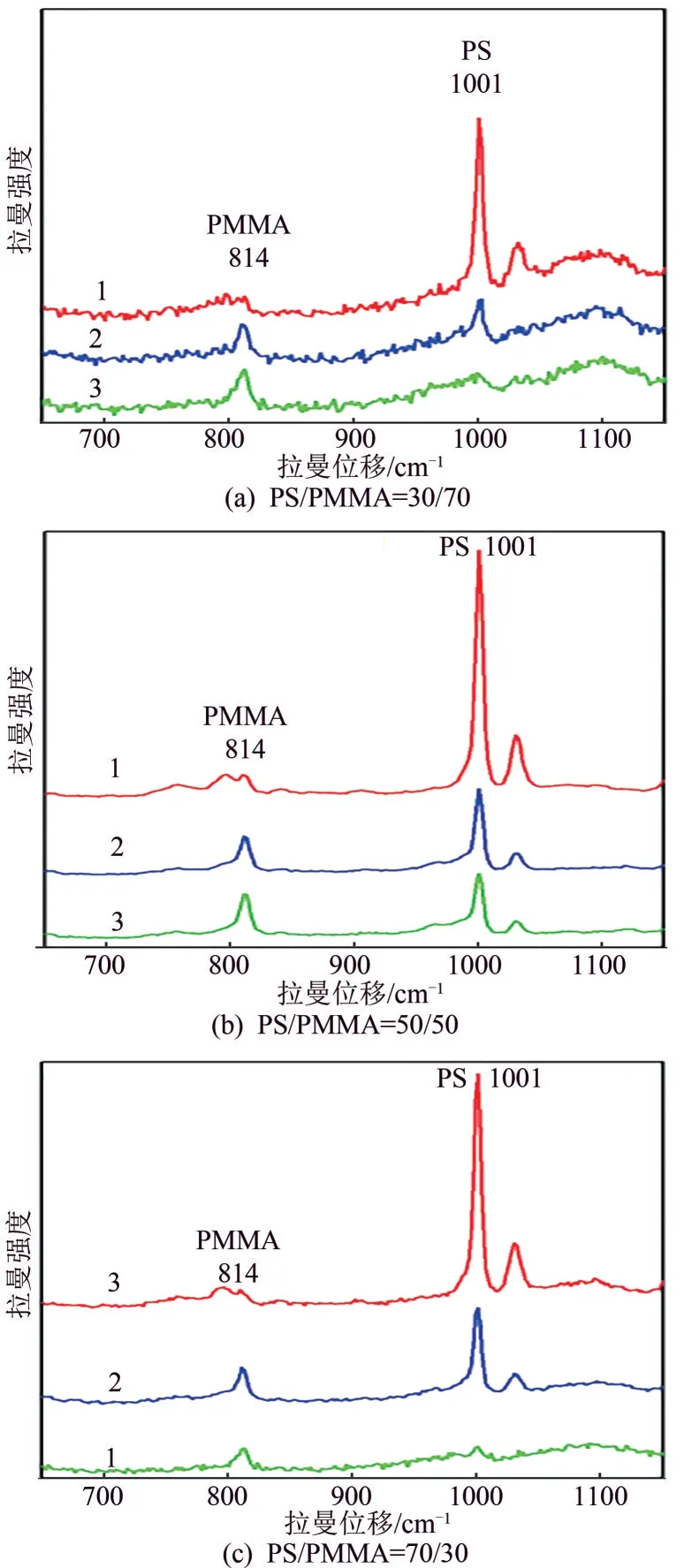
图4 图2(a2)、(b2)、(c2)中点1、2、3处的拉曼光谱
由上述分析可知,在PS/PMMA=70/30 共混体系中,采用光谱标定的方法可知PMMA 是分散相,PS 是连续相。为了进一步证明该结论,采用拉曼线性扫描分别对PS/PMMA=70/30 和PS/PMMA/PS-b-PMMA=70/30/5 共混体系进行了成分分析,结果如图5 所示。图5(a)上部分是PS/PMMA=70/30共混体系的光学显微图,图5(a)下部分是1001cm-1和814cm-1的拉曼散射强度随图5(a)上部分中彩色线条扫描步径的变化图,当激光经过分散相粒子上时,PS 特征峰1001cm-1的散射强度急剧下降,PMMA 特征峰814cm-1的散射强度增加,说明在体系中分散相是PMMA,连续相是PS,同时曲线的轮廓也反映出分散相粒子的大小。图5(b)上部分是PS/PMMA/PS-b-PMMA=70/30/5 共混体系的光学显微图,图5(b)下部分是1001cm-1和814cm-1的拉曼散射强度随图5(b)上部分中彩色线条扫描步径的变化图,曲线的变化趋势同PS/PMMA=70/30 共混体系一致,说明增容剂的加入不会使分散相和连续相发生相翻转,但是明显减小了分散相的粒子尺寸,分散相粒子的尺寸和分布变得均匀。PS 和PMMA 的成分比例变化如图6 所示,当PS 的含量减少时,PMMA的含量增加,曲线呈现互补形式,说明拉曼线性扫描可以有效地分析共混体系的组分构成。
众所周知,增容剂的加入可以降低共混体系的界面张力和提高分散度,从而提高向形态的稳定性。然而共混体系界面张力的降低不仅与增容剂的分子结构有关,而且与增容剂的浓度有密切的联系,如图7 所示。在PS/PMMA=30/70 共混体系中,增容剂PS-b-PMMA 的质量分数由0 增至10%,分散相PS 的粒径逐渐变小,其分布程度也越来越均匀,如图7(a)~(e)所示,平均粒径从7.5μm 下降至2.5μm,其Nc从0.26 增加到0.8,趋近于半相容性共混体系,说明增容剂的加入有利于改善PS 和PMMA相容性,同时增容剂的含量在不超过其表观临界浓度时,增容剂含量的增加可以进一步改善不相容体系的相容性[34]。
3 结论
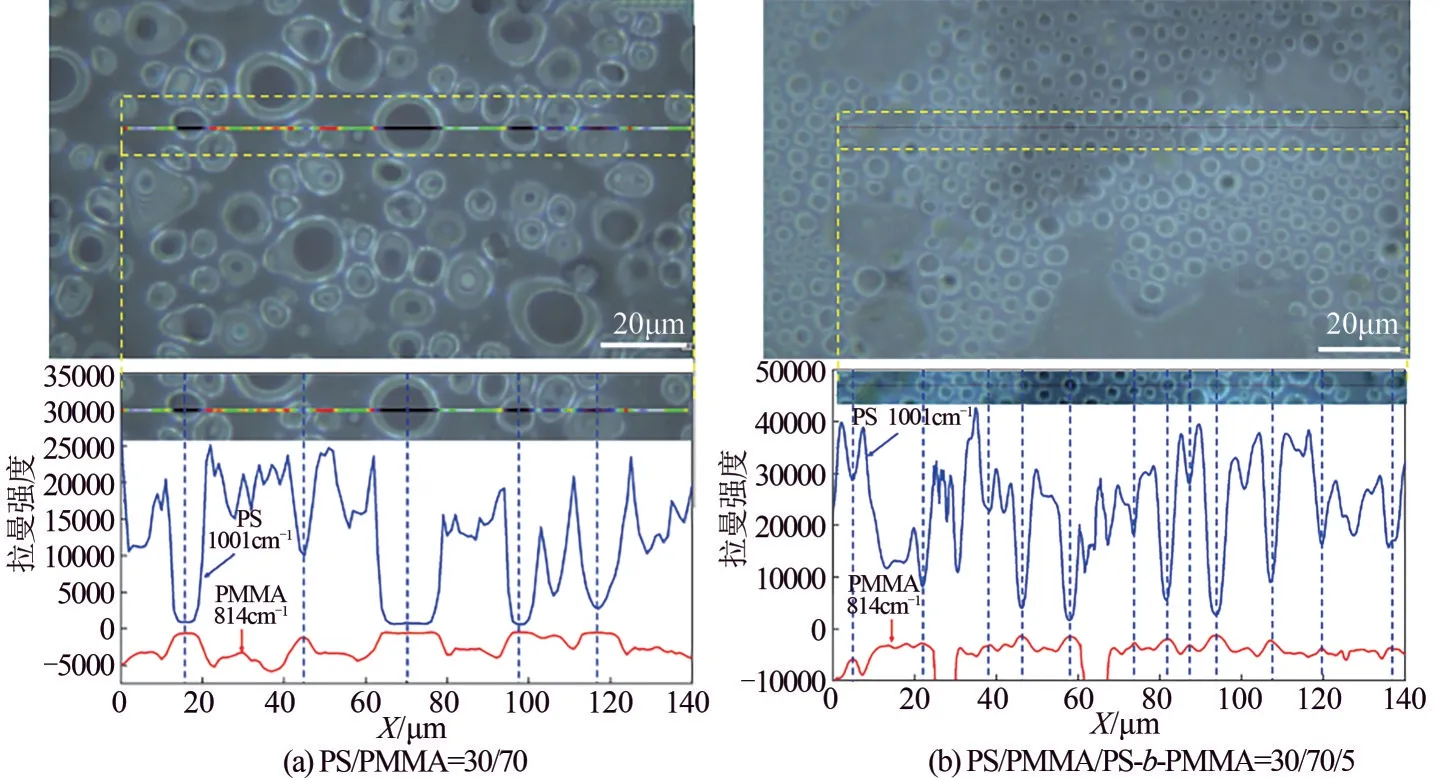
图5 共混体系PS/PMMA=30/70和PS/PMMA/PS-PMMA=30/70/5的光谱图及其谱带1001cm-1(PS的特征峰)和814cm-1(PMMA的特征峰)的拉曼散射强度在光学图彩色区域的强度分布图
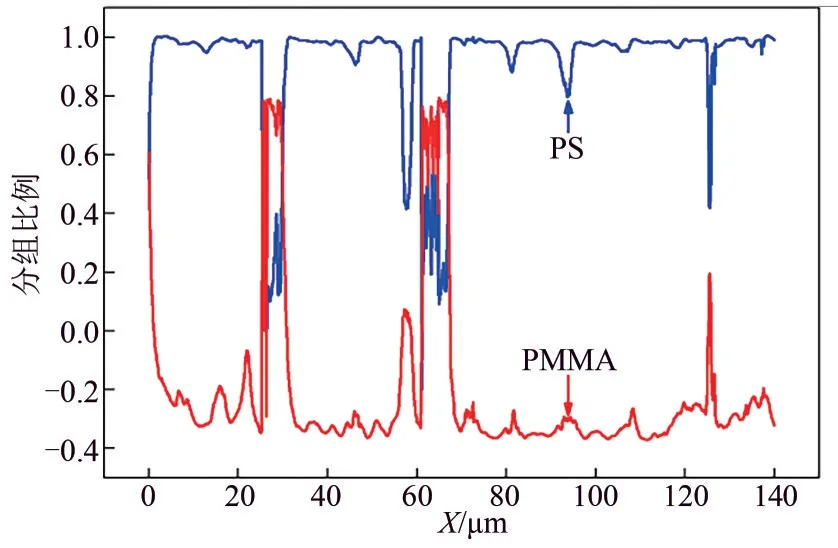
图6 共混体系中PS和PMMA随横向扫描步径的成分比例图(PS/PMMA=70/30)
本文采用溶液混合法制备了PS/PMMA 共混体系及其增容体系;采用原子转移自由基聚合的方法成功制备了PS-b-PMMA 嵌段共聚物,并将其作为PS 和PMMA 共混体系的增容剂。利用拉曼点到点扫描成像模式研究PS/PMMA 共混体系及其增容体系的相态结构及其相分布,研究发现共混体系的相态结构都以“海岛”形式存在。利用拉曼Mapping成像时的分子指纹光谱信息和线性扫描技术,获取了分散相和连续相的组分构成。当PS/PMMA=30/70 时,分散相为PS,连续相为PMMA;当PS/PMMA=50/50 时,分散相为PS,但PS 分子链仍存在于PMMA 连续相中;当PS/PMMA=70/30 时,分散相为PMMA,连续相为PS。在PS/PMMA 共混体系中加人PS-b-PMMA 作为增容剂,能有效改善体系的相容性,这是因为增容剂起到连接桥梁的作用,促使不相容的PS 和PMMA 结合在一起,降低界面张力,增大两相界面,以减少相分离的发生,进而得到稳定的共混聚合物。拉曼Mapping结构相图证实了加入PS-b-PMMA 增容剂能有效改善PS/PMMA 共混体系的界面黏合力,体系的相容性增加。

图7 共混体系PS/PMMA=30/70中加入不同质量分数增容剂PS-b-PMMA的拉曼光谱成像图及平均粒径和Nc与增容剂质量分数的关系
