多孔有机聚合物催化二氧化碳合成环状碳酸酯研究进展
2021-07-28陈亚举任清刚周贤太纪红兵
陈亚举,任清刚,周贤太,纪红兵,
(1 广东石油化工学院化学学院,广东茂名 525000;2 广东石油化工学院材料科学与工程学院,广东茂名 525000; 3 中山大学化学学院精细化工研究院,广东广州 510275)
自1750 年工业革命以来,不断增长的人口、密集型工农业活动、土地利用和森林破坏等因素都促使大气CO2含量的急剧增加。据国际能源署于2019 年发布的最新统计显示,全球2018 年排放至大气中与能源相关的CO2量已持续增加至331 亿吨[1]。大气中快速增长的CO2不仅导致全球变暖,而且威胁着人类赖以生存的环境。CO2不仅是温室气体的主要成分,同时也是一种储量丰富、清洁、安全和可再生的碳一资源[2-3]。开发将CO2转化为化工基础原料、燃料和高分子的化学过程,是一条能有效消除大气中CO2和缓解能源危机的重要途径[4]。然而,CO2分子极其稳定,其发生化学反应需要克服其热力学能垒和动力学惰性[2]。因此,较高的能量输入也直接增加了CO2的排放,成为以上策略实施一个主要制约因素。

图1 CO2参与的典型有机反应
得益于绿色化学的快速发展,有关利用CO2为原料通过构建C―C、C―N、C―O 和C―H 键合成化学品的反应得到了飞速发展(图1)[5-8]。其中,在催化剂作用下,CO2与环氧化物通过环加成反应制备环状碳酸酯的反应能够在温和条件下发生,反应的推动力主要来源于环氧化物三元环环张力的释放。除此之外,该反应具有100%的原子经济性,且所生成的环状碳酸酯是一种能保证长期稳定的化合物,相比于其他化学品更符合碳存储的要求[9]。同时,环状碳酸酯具有的广阔应用前景,可作为有机合成中间体、极性非质子溶剂、锂电池电解质和聚合物合成单体等[10]。
基于以上考虑,利用CO2合成环状碳酸酯是一条环境友好型且更加符合碳平衡策略的方法,特别是在直接利用低浓度CO2或工业废气时尤具优势[11]。为了实现这一反应的高效进行,至今为止已经开发了丰富多样的均相和多相催化剂,如金属卤化物[12]、金属氧化物[13]、离子液体[14-15]、有机碱[16]、金属配合物[17-22]、金属有机框架材料(MOFs)[23-24]和多孔有机聚合物(POPs)[25-26]等。其中,POPs 作为一种新兴的多孔材料,因其优异的物理化学性质,被广泛应用于气体吸附、分离、储能、新材料和催化等领域。特别在CO2的捕集和转化方面,POPs 高CO2吸附容量和吸附选择性以及优异的化学可调控性,在催化CO2参与的有机反应中潜力巨大,并且已取得了令人瞩目的成果[27]。本文基于POPs催化材料的合成方法、结构和组成特性层面,对近年POPs 催化CO2与环氧化物反应制备环状碳酸酯的研究进行综述。同时,对目前POPs 材料的开发及其在CO2吸附与转化领域的应用发展方向进行展望。
1 多孔有机聚合物
POPs 一般指有机基团或片段通过不可逆的化学反应聚合形成的无定形多孔材料。根据其形成方式和结构特点,主要可以分为固有微孔聚合物(polymer of intrinsic microporosity,PIMs)、共轭微孔聚合物(conjugated microporous polymers,CMPs)、超高交联聚合物(hyper-crosslinked polymers,HCPs)、多芳香骨架材料(porous aromatic frameworks,PAFs)、共价三嗪类骨架材料(covalent triazinebased frameworks,CTFs) 以及多孔聚合物网络(porous polymer networks,PPNs)等。
目前,合成POPs 的主要途径有Sonogashira-Hagihara 偶联反应、Yamamoto-Ullmann 偶联反应、Suzuki 偶联反应、Friedel-Crafts 烷基化反应、氰基环化反应、自由基聚合、席夫碱缩合反应、“点击”反应和酚醛树脂反应等化学反应。POPs 具有高比表面积、多孔性、低密度性、高化学物理稳定性以及聚合方法多样性。近年来,POPs 在CO2催化转化方面也取得了重要研究进展[28-29]。结合目前广泛认可的CO2与环氧化物合成环状碳酸酯的酸碱协同催化机理(图2)[8],以下将从POPs 中催化活性组分构成角度,分别综述其在该环加成反应的研究进展,包括金属配合物类、氢键供体和离子液体等一元活性组分基POPs体系以及金属配合物/离子液体、氢键供体/离子液体等二元活性组分基POPs体系。

图2 CO2与环氧化物环加成反应的酸碱协同反应机理
2 含一元活性组分的POPs催化剂
2.1 金属配合物基POPs催化剂
2.1.1 金属卟啉配合物类
首次将金属卟啉作为单体进行聚合反应得到相应的多孔有机催化剂,并应用于CO2与环氧化物的环加成反应是在2015 年。Wang 和Qin 课题组[30]以1,3,5-三苯乙炔和四溴苯基铝卟啉为原料,利用[Pd(PPh3)4]/CuI 催化的Sonogashira 聚合反应,制备了一种金属卟啉基共轭微孔聚合物Al-CMP(图3,1)。该微孔聚合物比表面积(BET)为839m2/g,孔径分布集中在1.42nm,在常温常压条件下,其表现出较高的CO2吸附量(27.43mg/g)。在催化CO2与环氧化物生成环状碳酸酯的反应中,当反应条件为100℃、3.0MPa CO2、0.05%(摩尔分数)的Al-CMP催化剂和0.5%(摩尔分数)的双(三苯基正膦基)氯化铵助剂时,反应5h 后,环氧丙烷(PO)的转化率可达91%,相应的TON(单位活性位的转化数)值为1820,展现了该催化剂的良好催化活性。同年,Chen等[23]利用经典的酸催化氧化法,以吡咯和对苯二甲醛为原料原位聚合,生成具有三维网络结构的卟啉基有机聚合物。接着对上述卟啉配体进行金属化,最终获得了两种金属卟啉基POP材料,分别为M(Por)OP(M=Zn,Co)(图3,2)。该类聚合物也成功应用于上述环加成反应中。催化剂反应6次后,其活性和选择性没有显著下降,表明其在该体系中具有良好的稳定性和重复使用性。随后,该课题组[31]在上述金属卟啉基POP材料的基础上,在合成过程中加入适量的二氧化硅涂覆的磁性纳米粒子(Fe3O4@SiO2),在原位形成聚合物的同时,将Fe3O4@SiO2纳米颗粒包覆进聚合物纳米孔道中,从而赋予该类聚合物以磁性,所获得的磁性金属卟啉基聚合物Fe3O4@SiO2@Zn(Por)OP 在催化CO2合成环状碳酸酯的反应中依然保持良好的催化性能,与之前的工作相比,可以更简捷地实现聚合物回收。
鉴于金属卟啉配合物基POPs 催化剂在CO2环加成反应中表现出的优异性能,越来越多的科学家通过设计金属卟啉配合物单体,利用多样化的聚合反应开发出一系列该类多相催化剂。其中,Xiao等[32]通过经典的自由基聚合反应,以偶氮二异丁腈为引发剂、四乙烯基苯基卟啉为原料,高效合成了高比表面积的卟啉基多孔聚合物(POP-TPP),利用钴盐、镁盐和锌盐成功对其进行了金属化,获得了一系列金属卟啉基POP 材料(M/POP-TPP,M=Co、Zn、Mg)(图3,3)。该作者利用氮气吸脱附实验得出,POP-TPP 和Co/POP-TPP 的比表面积(BET)分别可达到1200m2/g 和924m2/g,具有丰富的多级孔结构(孔径分布于0.4~2nm 和12~150nm)。此外,在其常温常压下CO2吸附实验中得出,POP-TPP 和Co/POP-TPP 的CO2吸附量分别为29.7cm3/g 和32.6cm3/g。随后,该作者将M/POPTPP 催化剂成功应用于以四丁基溴化铵(TBAB)为助剂的CO2/环氧氯丙烷环加成反应中。在29℃和0.1MPa 条件下,反应24h 其转化率可达95.6%,并且无副产物生成。值得注意的是,在以体积分数分别为15%和85%的CO2为原料时,相同条件下多相Co/POP-TPP体系表现出比均相Co-TPP体系更优的催化性能。为此,该作者认为是Co/POP-TPP 具有丰富的纳米孔道结构,而这些孔道有利于CO2的吸附,使得CO2富集于孔道中的催化活性位点附近,进而加速了反应的进行。2018 年,Wang 和Ding等[33]也利用类似的方法,将四乙烯基苯基镁卟啉与二乙烯基苯共聚,得到了一系列具有良好溶胀性能的多孔有机聚合物,最高比表面积可达2077m2/g。
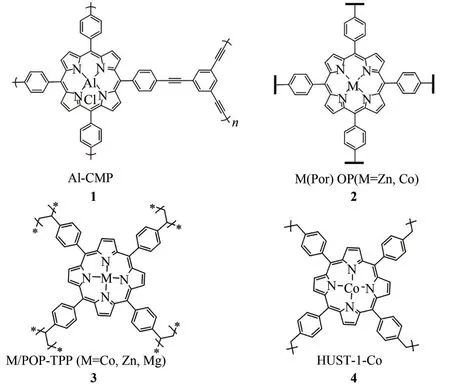
图3 几种金属卟啉配合物基多孔聚合物的结构示意图
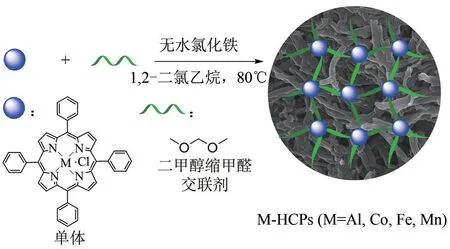
图4 Friedel-Crafts反应合成M-HCPs(M=Al,Co,Fe,Mn)示意图[35]
随后,Tan 课题组[34]以四苯基卟啉为原料,通过以氯化铝为催化剂的Friedel-Crafts反应,获得了超交联卟啉基聚合物。进而,利用后上金属的方法制备得到HUST-1-Co(图3,4)。该聚合物比表面积(BET)为1360m2/g,CO2吸附量为13.17%(质量分数,1bar 和25℃,1bar=105Pa)。Ji 和Luo 等[35]相继报道了利用Friedel-Crafts烷基化反应,将催化剂替换为更容易进行后处理的无水氯化铁,以二甲醇缩甲醛为交联单体、四苯基金属卟啉为原料,一步合成了一系列不同金属中心的金属卟啉基超交联聚合物M-HCPs(M=Al,Co,Fe,Mn)(图4)。该类聚合物同样具有高比表面积、丰富的纳米孔道、良好的CO2吸附性能和CO2/N2选择吸附性能及热稳定性。此外,通过透射电镜表征,发现该聚合物为中空管状结构。在上述合成环状碳酸酯的反应中,Al-HCP 表现出优异的催化性能,并且超过了其均相单体Al-TPP 的性能。在条件为40℃和1.0MPa时,加入0.25%(摩尔分数)的Al-HCP和2%(摩尔分数)的TBAB,仅需反应1h,PO 完全转化为相应的环状碳酸酯。基于以上结果,该作者进一步将温度提高至100℃,[PO]/[Al-HCP]降低至1∶20000,依然可以获得高达14880h-1的初始TOF值,这在当时属于最高值之一。同样,该类催化剂可简单回收并且稳定重复使用10 次以上。最后,该作者对此类超交联聚合物在此反应中表现出的优异性能的可能原因进行了分析:①Al 具有较强的Lewis 酸性;②催化剂框架中的Al位点含量丰富且分散均匀,有利于底物与活性位点的接触;③纳米孔道具有富集CO2的作用,同时中空结构有利于底物和产物的扩散,进而提升了该反应的动力学行为。
通过对上述文献分析可知,在金属卟啉基POP催化剂中引入多级孔结构,可以充分发挥催化剂框架中微孔对二氧化碳的富集作用以及介孔对反应物和产物分子的扩散强化作用。进一步,科学家在合成该类催化剂的过程中直接引入硬模板剂,用于后续制造大量的介孔。Son等[36]以四(4-乙炔基苯)卟啉和对二碘苯为原料,在进行Sonogashira偶联反应的同时,加入一定量的SiO2纳米球。经过聚合、酸刻蚀和金属化等步骤后,得到了具有中空结构的球状超交联聚合物(MCrPN和MZnPN)。该类聚合物在常温常压条件下可有效催化各种环氧化物与CO2反应合成相应的环状碳酸酯,这也印证了多级孔结构对该反应过程强化具有的重要作用。同时,该作者也详细考察了不同助催化剂,如TBAB(四丁基溴化铵)、TBAI(四正丁基碘化铵)、TBACl(四正丁基氯化铵)、TNAB(四正己基溴化铵)、TOAB(四正辛基溴化铵)和PPNCl[双(三苯基正膦基)氯化铵]对该反应的影响,为后续关于助催化剂的筛选提供了一定指导作用。
2.1.2 金属Salen配合物类
2013 年,Deng 课题组[37]首次报道了通过Sonogashira 偶联反应而得到的金属Salen 配合物基CMP 材料(Co-CMP 和Al-CMP)(图5,5)。其中Co-CMP的比表面积高达965m2/g,常温常压下CO2吸附量为79.3mg/g。随后该类多孔材料成功应用于催化以TBAB 为助催化剂的CO2/环氧化物环加成反应,并且在重复使用22 次后,其活性和选择性没有明显变化。这类催化剂也实现了常温常压下高选择性合成环状碳酸酯,展现了该类有机多孔材料在CO2捕集与转化方面的巨大应用潜力。此后,该课题组陆续报道了类似的CMP 材料(Zn-CMP 和Cr-CMP)[38-39]。其中,在120℃和3.0MPa 条件下,以0.0025%(摩尔分数)的Zn-CMP 为催化剂和0.9%(摩尔分数)的TBAB 为助剂,该反应的初始TOF值高达11600h-1。2013年,Son等[40]将1,3,5-三乙炔苯替换为四(4-乙炔基苯)甲烷,同样利用Sonogashira-Hagihara偶联反应制备得到一系列金属Salen配合物基多孔聚合物(Co-MON、Al-MON 和Cr-MON)(图5,6)。2016 年,Eddaoudi 等[41]利用相同的聚合方法,获得了类似的多孔聚合物材料1-Co 和1-Cr。以上这些聚合物在CO2和环氧化物合成环状碳酸酯的反应中也表现出良好的催化性能和重复使用性。

图5 几种金属Salen配合物基多孔聚合物的结构示意图
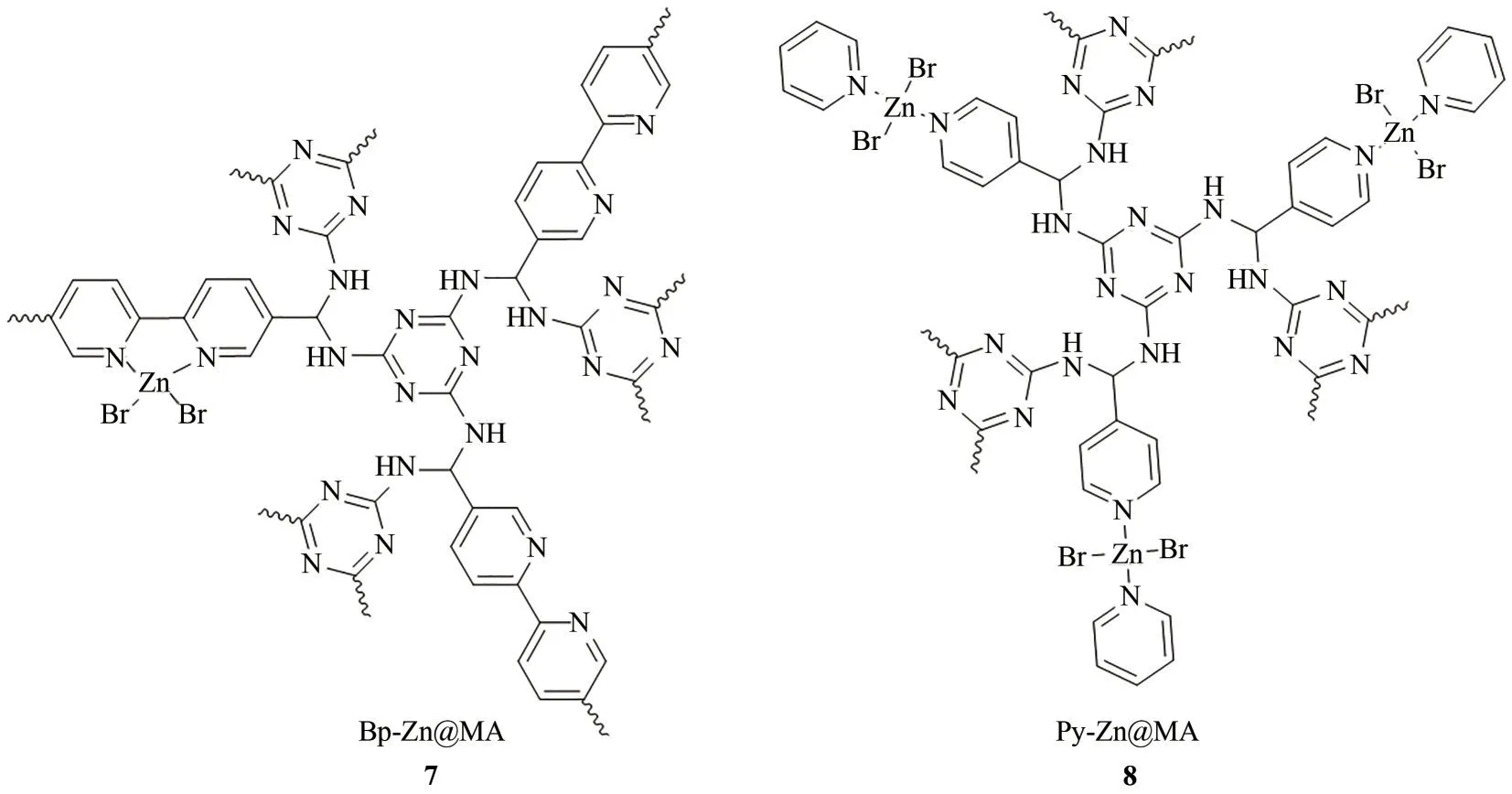
图6 Bp-Zn@MA和Py-Zn@MA的结构示意图
随后,Bhaumik 等[42]以3,3-二氨基联苯胺、2,4-二甲酰基间苯三酚和二水合醋酸锌为原料,通过原位生成Salen Zn结构,一步合成了具有高比表面积(423m2/g)的多孔聚合物Zn@SBMMP。当以TBAB 为助剂、二氯甲烷为溶剂时,该聚合物催化剂可高效高选择性地催化环状碳酸酯的合成,并且表现出良好的底物扩展性。这种定向原位构建功能单元的方法,也为合成功能型POPs 提供了一条原子经济性和简捷的合成策略。
2.1.3 其他金属配合物类
2016 年,Yang 等[26]以醛基功能化的联吡啶锌配合物和三聚氰胺为原料,在溶剂热条件下聚合得到具有多级孔结构的聚合物Bp-Zn@MA(图6,7)。该材料用于催化以TBAB 为助剂的CO2环加成反应,当反应温度升高至150℃、CO2压力为1MPa时,反应1.5h即可获得>99%的转化率和96%的选择性,相应的TOF 值可达8041h-1,这一值在该反应多相催化体系中十分难得。随后,该课题组[43]利用类似的溶剂热聚合方法,获得了含有吡啶锌配合物单元的多孔聚合物Py-Zn@MA(图6,8)。该聚合物具有丰富的多级孔结构,其比表面积(BET)为207m2/g,常温常压下CO2吸附量为1.33mmol/g。该聚合物可高效高选择性地催化无助剂和无溶剂条件下的CO2/环氧化物环加成反应。此外,文献作者将稀释的CO2(20%CO2+80%N2,体积分数)作为原料,该催化剂依然表现出良好的催化性能和重复使用性。这一结果对将工业废气转化为高附加值的化学品起到一定的基础研究意义。
2016 年,Zhang 等[25]以3,3-二氨基联苯胺为原料,在醋酸碘苯的氧化偶联作用下,形成了以偶氮基键联的多孔聚合物PAF。通过氮气和CO2吸脱附测试,该聚合物的比表面积高达742m2/g,CO2吸附量为11.9%(25℃,1bar)。文献作者利用热重分析法,对该材料进行持续可逆吸附CO2循环8 次(500min)测试,结果表明其具有优异的可逆吸附性能。考虑到框架中含有丰富的氮原子,文献作者进行了CO2/N2选择性吸附实验,获得了较高的CO2/N2选择吸附比值(42∶1,25℃),表明该类聚合物具有一定的CO2/N2分离能力。随后,将一定量的ZnBr2通过络合作用负载于PAF框架中,得到PAFZnBr2。在催化CO2环加成反应中,加入助剂TBAB,PAF-ZnBr2表现出良好的催化性能、底物扩展性和稳定性。
2017年,Xiao等[44]利用自由基聚合反应开发了典型的金属联吡啶配合物基POPs,如图7 所示。该类聚合物具有较高的比表面积以及丰富的多级孔,且在CO2吸附性能方面也有着良好表现。在用作催化剂时,能够在室温和常压(29℃和1bar)条件下实现低浓度CO2(15% CO2+85% N2,体积分数)高效转化为环状碳酸酯,并且具有良好的循环使用性。文献作者指出,在该反应体系中,催化剂微孔有利于将CO2富集于金属活性位点M(M=Cu,Zn,Co)附近,而介孔结构有利于底物和产物分子扩散。该工作也对设计具有高效催化性能的多孔催化材料具有一定的指导意义。
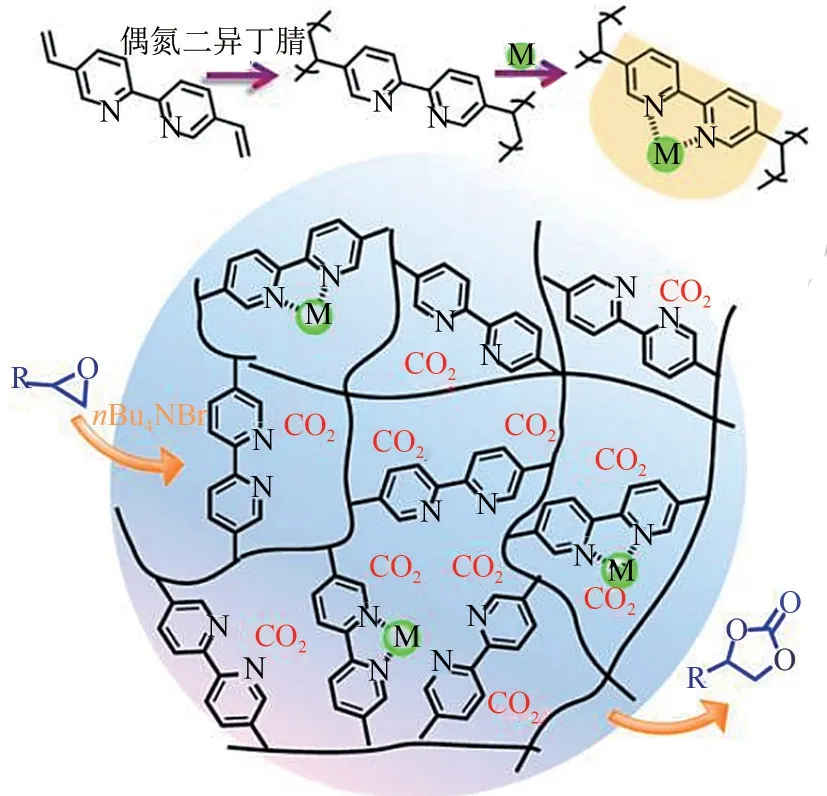
图7 M/POP-Bpy的合成及其在CO2转化中的应用[44]
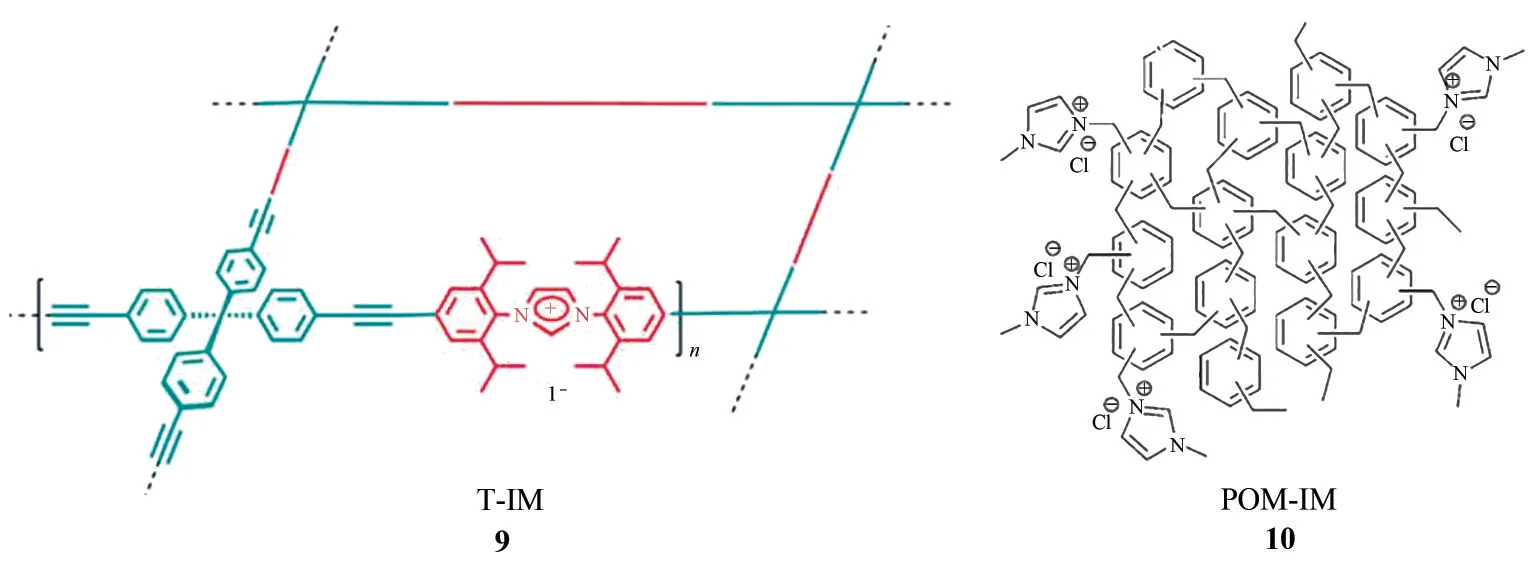
图8 T-IM和POM-IM的结构示意图
2.2 离子液体基POPs催化剂
2.2.1 咪唑离子液体类
早在2011 年,Son 课题组[45]就利用Sonogashira偶联反应成功获得了具有三维网络结构的咪唑离子液体基多孔聚合物T-IM(图8,9)。该聚合物为典型的微孔材料,其比表面积(BET)为620m2/g,并且呈现出管状结构(外径2.0μm±0.4μm,壁厚290nm±50nm)。框架中含有的丰富的卤素阴离子(I-)可作为亲核试剂进攻环氧化物,形成有利于CO2插入的开环中间体。使用0.065%(摩尔分数)的T-IM 为催化剂,在160℃和3MPa 条件下反应10h,可获得92%的4-氯甲基碳酸乙烯酯收率。
上述偶联反应,除了单体合成过程复杂之外,贵金属催化剂的使用也使得聚合物纯化难度加大和制备成本增加。因此,探索其他更加符合绿色化学理念的聚合策略更具吸引力。为此,Zhang 等[46]首先利用Friedel-Crafts烷基化反应合成了含有丰富氯甲基或溴甲基的超交联聚合物。再利用后合成法(post-synthetic method),将咪唑离子液体嫁接于超交联聚合物框架中,获得了一系列咪唑离子液体基超交联聚合物POM-IM(图8,10)。该类聚合物具有大量的纳米孔道,其比表面积(BET)最高可达到926m2/g,CO2吸附量可达14.5%(0℃,1bar)。在120℃、1MPa 和2mL 乙醇为溶剂的条件下,以POM-IM化合物为催化剂,反应8h,可获得94%的PO 转化率。考虑到获得POM-IM 催化剂采用的是后修饰策略,该策略可能会由于催化剂框架中反应位点在嫁接过程中反应不完全而导致离子液体活性组分含量不高,同时外围聚集的离子液体单元也会对孔道产生阻塞作用。科学家们利用Friedel-Crafts(傅-克)烷基化反应与碱化反应合成了一系列超高交联聚合物,在聚合过程中伴随生成了咪唑离子液体,因此得到了超高交联离子液体聚合物(图9)[47]。这类聚合物具有高比表面积(>534m2/g)和高CO2吸附量(>2.4mmol/g,273K,1bar)。当离子液体中的阴离子为Br-时,在120℃和10bar 下反应6h,环氧苯乙烷几乎完全转化为目标产物。当加入Lewis 酸ZnBr2作为助剂时,催化性能大大提高,在常温常压下就能实现该反应高效高选择性地进行,只是反应时间需要延长至120h。同样,利用15%的CO2(在85%N2中,体积分数)为原料,也能够实现环状碳酸酯的顺利合成。最近,Huang等[48]也利用该类反应获得了类似多孔离子聚合物催化剂,也成功应用于常压条件下催化CO2合成环状碳酸酯的反应中。
2015 年,Bordiga 等[49]以双键功能化的咪唑和二乙烯基苯(DVB)为单体,在自由基引发剂偶氮二异丁腈(AIBN)的作用下,合成了一系列含丰富咪唑基团的聚合物。利用卤代烃对咪唑进行离子液体化,并进行阴离子交换,获得了一系列咪唑离子液体基多孔聚合物,也成功应用于CO2/环氧化物的环加成反应中。2015 年,Wang 等[50]设计了一系列双键修饰的咪唑离子液体单体,通过自由基聚合反应,并在聚合过程中引入少量水,在此过程中水与极性的IL 基团相互作用,使得聚合后离子液体具有较高的比表面积(205m2/g)。多孔性使得该离子液体聚合物在常压下具有更高的CO2吸附量(1.02mmol/g),这有利于CO2分子在催化活性中心富集,同时多孔结构也加速了反应物与产物的传质,因此催化效率或会很好地提升。在1.0MPa 和110℃下,只需2h,环氧氯丙烷几乎完全转化为目标产物。而在常压70℃条件下,延长反应时间至48h,也能获得99.4%的转化率,且几乎无副产物生成。最近,Wang 等[51]利用叠氮基和炔基发生的“点击”反应,制备得到一系列富氮POP(CPP),并成功应用于CO2的环加成反应中。由于大体积和大位阻的四苯基甲烷和四苯基乙烯结构单元的引入,在较低咪唑离子液体含量时,该类聚合物具有较大的比表面积(994m2/g)。最近,Gai等[52]以氯化乙醚双(1-偏二咪唑)与乙二醇二甲基丙烯酸酯为原料进行共聚合,制备了一系列结构新颖的聚离子液体,该类聚离子液体比表面积最高只有22.8m2/g。尽管如此,由于其具有丰富的催化活性中心,在CO2的环加成反应中依然表现出良好的活性,在100℃和1MPa下反应6h,可获得99.7%的碳酸环氯丙烯酯收率。然而,随着离子液体含量的增加,比表面积急剧下降,这也是当前制备离子液体多孔材料面临的普遍问题。因此获得高比表面积和高离子液体含量的多孔离子聚合物仍然是一项巨大挑战。

图9 HIPs的合成[47]
2.2.2 季鎓盐类
早在2012 年,Zhang 等[53]通过Yamamoto 偶联反应,以具有刚性结构的溴化四(4-氯苯基)季鏻盐为单体,合成了具有高比表面积(650m2/g)的季鏻离子液体聚合物。该聚合物具有丰富的微孔和少量的介孔。在合成环状碳酸酯的反应中,其表现出了良好的催化性能和重复使用性。2015 年,Zhang 课题组[54]利用Friedel-Crafts 烷基化反应,将一系列季鏻盐与交联剂FDA 聚合生成一系列季鏻型超交联聚合物(图10)。特别指出的是,文献作者通过加入少量的苯作为聚合单体,有效地克服了聚合过程中季鏻盐单体苯环缺电子效应的影响。该类聚合物比表面积最高可达1168m2/g,也表现出较好的CO2吸附性能(4.8%~7.5%,25℃和1bar)以及良好的CO2/N2选择吸附性(吸附比值为28~56,25℃和1bar)。在催化CO2与环氧化物合成环状碳酸酯的反应中,反应条件为DMF (2mL)、CO2(1MPa)和130℃,以ZnBr2为助剂时,反应4h 可获得94%的PO转化率和90%的碳酸丙烯酯(PC)收率。
2016年,Ma和Meng等[55]采用双键功能化的季鏻盐为单体,用自由基共聚法也获得了高比表面积(>402m2/g)的聚合物催化剂PIPs,相比于之前报道的类似聚合物合成方法,该策略更加绿色和高效。文献作者将该聚合物应用于催化无溶剂条件的CO2/环氧氯丙烷环加成反应中,在100℃和1bar 条件下,反应3h 后几乎所有的底物均转化为相应的环状碳酸酯。当进一步降低反应温度至室温时,通过延长反应时间至80h,也能够获得94.7%的目标产物收率。此外,该类催化剂在循环使用10次后,其活性和选择性没有明显降低。
随后,Zhao等[56]报道了以三烯丙基胺为原料制成的双键修饰的离子液体HTA/BTA,与1,4-二乙烯基苯利用AIBN 引发共聚形成有机多孔材料(DVB-HTA 和DVB-BTA)。该材料催化环氧丙烷与CO2环加成反应转化率可达到90%以上。其催化活性相对其他离子液体来说比较高。重复使用4次后,其活性几乎无降低,红外检测显示其结构几乎没有变化。近期,Li 和Lu 课题组[57]也利用以偶氮二异丁腈为引发剂的自由基聚合反应合成了具有海绵性状的季铵类多孔离子液体聚合物(图11)。在聚合反应完成后,所获得的MPILs为凝胶状,文献作者通过不同的干燥方式,包括超临界二氧化碳干燥、真空干燥及常压干燥,对PDBA-Cl进行干燥,获得的样品分别为PDBA-Cl-SCD、PDBA-Cl-VD和PDBA-Cl-AD。通过77K条件下的氮气吸脱附实验对这些聚合物进行了测试,PDBA-Cl-SCD 和PDBA-Cl-VD 的比表面积(BET)分别为211.0m2/g和59.6m2/g,而通过常压条件下干燥获得的PDBACl-AD几乎没有比表面积。由此可以看出,通过超临界二氧化碳干燥法处理得到的样品具有最高的比表面积,该作者认为超临界流体近乎零表面的张力,使得在干燥过程中去除溶剂时,纳米孔道结构得到很好的保持,这也为今后处理这类样品提供了经验。在90℃和1bar 条件下,以PDBA-Cl-SCD 为催化剂,反应6h 后环氧氯丙烷完全转化为相应环状碳酸酯。
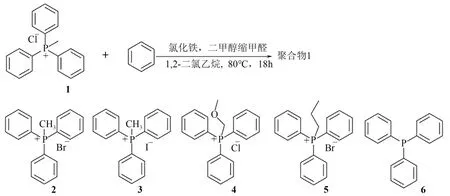
图10 季鏻盐类多孔有机聚合物的合成

图11 海绵状季铵盐类聚合物的合成及其在CO2/环氧化物环加成反应中的应用[57]
2.2.3 吡啶离子液体类
2016年,Coskun课题组[58]报道了一种具有立体网络结构的共轭联吡啶离子液体基多孔聚合物。该类聚合物是通过Sonogashira-Hagihara 偶联反应,以碘苯功能化的联吡啶离子液体和四(4-乙炔基苯)甲烷为原料聚合得到的。通过阴离子交换获得了一系列多孔材料PCP-Cl、PCP-BF4和PCP-PF6,这些聚合物为典型的微孔材料,比表面积分别为755m2/g、586m2/g 和433m2/g。由于样品中含有丰富的亲CO2离子基团和氮原子,文献作者对其进行了CO2吸附和CO2/N2选择吸附测试。以比表面积最高的PCPCl 为例,其在常温常压条件下的CO2吸附量为61.4mg/g。同时,CO2/N2选择吸附比为34∶1。随后,该作者考察了PCP-Cl 在CO2环加成反应中的催化性能,在90℃和1bar条件下,反应12h后环氧丙烷均转化为目标产物。
Leng 等[59]以1,2,4,5-四溴甲基苯和4,4'-联吡啶为原料,通过简单的离子化反应即获得了吡啶离子液体基聚合物。文献作者考察了不同溶剂对所形成聚合物形貌的影响,在四氢呋喃、甲苯和乙腈中获得的样品分别为TBB-Bpy-a、TBB-Bpy-b 和TBBBpy-c。通过扫描电镜表征,这三种样品均表现为较光滑的球状形貌,而以乙酸乙酯为溶剂获得的TBB-Bpy-d 则表现为无定形网络结构。以TBBBpy-a 为例,该作者进行了77K条件下的氮气吸脱附表征,其比表面积为10m2/g,低的比表面积也与扫描电镜所表征的光滑表面相一致。尽管该类聚合物的比表面积很低,但在催化CO2环加成反应中,依然表现出良好的催化活性和稳定性。120℃和1MPa 时,反应4h 后,可获得99%的PO 转化率,且无副产物生成。
2017年,Coskun课题组[60]以氰基功能化的联吡啶离子液体为原料、ZnCl2为催化剂,在熔融条件下获得了一类具有三嗪骨架的吡啶离子液体基多孔聚合物。虽然合成过程中条件苛刻,但是所得的离子聚合物cCTFs具有优异的热稳定性。不同温度条件下制备的cCTFs 分别为cCTF-400、cCTF-450 和cCTF-500,其比表面积分别为744m2/g、861m2/g和1247m2/g。比表面积这一递增趋势表明,升高反应温度有利于形成更高孔隙率的结构。相比其他CTF材料,在当时cCTF-500 具有最高的比表面积。以cCTF-500 为例,在25℃、1bar 和0℃、1bar 条件下的CO2吸附量分别为133mg/g 和80mg/g。该类聚合物也被应用于催化CO2/环氧化物环加成反应中,并表现出良好的催化活性和稳定性。
2.3 其他含一元活性组分的POPs催化剂
早在2012年,Roeser课题组[61]以氰基吡啶类化合物为原料、ZnCl2为催化剂,在熔融条件下首次获得了含有丰富碱性N位点的CTF材料。其中,在600℃条件下获得了无定形高比表面积的微介孔材料CTF-1-HAS(2087m2/g)和CTF-P-HAS(1745m2/g)。无溶剂条件下,以CTF-P-HAS为催化剂,130℃和6.9bar 下反应4h 即可获得100%的环氧氯丙烷转化率和95.8%的产物选择性。2015年,Coskun等[62]基于“点击”反应报道了一种卡宾类有机多孔聚合物NP-NHC。该聚合物是一种孔径(0.4nm)分布均一的微孔材料,其比表面积可达475m2/g,常温常压下的CO2捕获效率高达97%。基于卡宾类化合物可作为活化CO2的高效有机催化剂,该作者将该材料应用于CO2与环氧化物的环加成反应中。结果表明,在120℃和0.1MPa 条件下,NP-NHC 能够高效选择性地催化该反应进行。由于该材料孔径分布较窄,导致大体积反应物分子不能进入孔道而难以与活性位点发生接触,故催化活性极低,表现出择形催化效应。

图12 HF-MOP和PRP-1的结构示意图
2016年,Zhao等[63]报道了通过席夫碱缩合反应形成的富含酚羟基和亚胺键的微孔聚合物HF-MOP(图12,11),其比表面积为500m2/g。在催化以TBAI 为助剂的环加成反应中,酚羟基可作为氢键供体(HBD)与环氧化物中的氧形成氢键,同时TBAI 中的I-发生亲核进攻,使得环氧化物开环。由于上述协同效应,该体系在较温和条件下(80℃、2MPa)即可高效催化该反应进行。同年,Jiang课题组[64]采用溶剂热条件下的酚醛树脂缩合反应,同样获得了高比表面积(835m2/g)、富含酚羟基的多级孔材料PRP-1(图12,12)。该聚合物在常温常压条件下CO2吸附量可达71mg/g,并且表现出优异的CO2可逆吸附性能。上述环加成反应中,以PRP-1 为催化剂、TBAB 为助剂、乙腈为溶剂,在60℃和0.1MPa条件下反应24h,环氧氯丙烷的转化率为89%,且无副产物生成。此外,催化剂重复使用6次后,其催化活性和选择性无明显变化。最近,Yu 和Hu 等[65]以三聚氰胺和羟基苯甲醛为原料,在溶剂热条件下成功制备了一系列富含氮原子和酚羟基的介孔材料MOPs。该材料也成功应用于无溶剂、无助剂和温和条件下制备环状碳酸酯的反应中,并表现出良好的催化活性和循环使用性能。
最近,Song和Liang等[66]以倍半硅氧烷(POSS)和咔唑为原料,通过Friedel-Crafts烷基化反应制备了一系列孔隙率可控和荧光性质良好的介孔聚合物(SCHPPs)。研究表明,SCHPPs 具有高比表面积(799m2/g)和良好的热稳定性。SCHPPs 可作为非金属多相催化剂协同TBAB催化CO2的环加成反应。反应过程中催化剂咔唑单元中的仲胺作为氢键供体,通过与环氧化物的氧形成氢键而活化环氧化物,进而促进后续开环和CO2插入过程进行。该工作不仅促进了POSS 基多孔聚合物催化剂的开发,而且体现出该类材料在CO2环加成反应和发光探针方面的巨大应用潜力。Tao 等[67]将设计的乙烯基功能化离子液体单体与二乙烯基苯共聚得到了一系列多孔离子聚合物,该类聚合物在纯CO2以及低浓度CO2(体积分数15%,在N2中)的环加成反应表现出优异的催化活性。
3 含二元活性组分的POPs催化剂
基于以上关于单一活性中心的POPs 催化剂的总结,离子液体、金属配合物或氢键供体(HBD)构成的POPs 催化剂在环氧化物与CO2的环加成反应中具有高效性。但该类催化剂若想在温和的反应条件下获得优异的催化性能,往往需要在催化体系中同时引入酸(金属配合物或氢键供体)和碱(离子液体)两种活性位。从上述反应体系中可以看出,其中一种活性组分直接构筑于POPs框架中时,另一活性组分则是以助剂的形式加入。如此一来,均相助剂的引入也导致后续产物分离和纯化的难度加大,使成本大大增加。显然,将Lewis 酸金属配合物或者具有氢键供体(HBD)的羟基、羧基和离子液体等位点同时引入多孔聚合物中,便形成了包含多种活性组分的POPs 催化材料,即可很好地解决上述问题。为此,科学家们在这方面展开了大量研究。
3.1 金属卟啉配合物/离子液体类
2017 年,Ji 和Luo 等[68]首次报道了采用经典的Yamamoto-Ullmann 偶联反应,在双(1,5-环辛二烯)镍的催化作用下,合成了一系列金属卟啉基离子液体多孔聚合物催化剂(Al-iPOP-1 和Al-iPOP-2)(图13)。这种合成方法所使用的催化剂在反应结束后,可以通过无机酸简单处理而除去,比传统的钯系催化剂更具优势。该类聚合物具有较高的比表面积、良好的CO2亲和性和CO2/N2选择吸附性。尤其该类双功能聚合物还具有优异的溶胀性能,可提供有益于反应的微环境。因此,在更加温和的条件下(40℃,1.0MPa),以上多孔催化剂高效高选择性地实现了环状碳酸酯的合成。当反应温度升高至100℃,在0.01%(摩尔分数)Al-iPOP-2的催化作用下,该反应的初始TOF 值高达7600h-1。此外,该催化剂也能实现低浓度CO2捕集与高效催化转化为环状碳酸酯,且展现出良好的稳定性和重复使用性。

图13 Al-POP、Al-iPOP-1和Al-iPOP-2的合成[68]
随后,该课题组等[69]采用“自下而上”合成策略,在溶剂热条件下,通过席夫碱缩聚反应,成功制备了一系列同时含有金属卟啉和离子液体单元的多孔有机材料(SYSU-Zn@IL-1和SYSU-Zn@IL-2)。灵活的框架、丰富的极性基团和杂原子,赋予该类聚合物在常温常压下具有较强的CO2亲和作用,并获得了较高的CO2吸附量和CO2/N2吸附选择性。由于具有路易斯酸性的金属中心与具有亲核性的溴离子之间的协同催化作用,该类双功能催化剂实现了温和条件下(80℃,1.0MPa)催化CO2/PO 环加成合成环状碳酸酯。此外,该类多相催化剂还表现出优异的稳定性、良好的重复使用性和底物普适性。以模拟废气(体积分数15%CO2,在N2气氛中)为原料时,环氧化物仍可顺利转化为目标产物。
2018年,Ding课题组[70]利用自由基共聚反应将咪唑离子液体和金属卟啉构筑于同一聚合物框架内,得到双功能多孔离子液体聚合物Mg-por/pho@POP(图14,13),实现了温和条件下协同催化该环加成反应高效进行。而在140℃和1.0MPa CO2下,反应初始TOF 值高达15600h-1。同年,Yang 课题组[71]将聚合反应和原位涂覆技术相结合,使得聚合形成金属卟啉基离子液体的同时涂覆于碳纳米管表面,得到一种多功能的离子液体聚合物催化剂ZnTPy-BIM4/CNTs(图14,14)。碳纳米管的引入增大了聚合物比表面积,使得反应底物与活性位点接触面积增大,更有利于卟啉金属中心和卤素阴离子的协同催化。同时碳纳米管的纳米孔道有利于反应物和产物的传质,因而该催化剂表现出优良的催化活性和稳定性。
3.2 金属Salen配合物/离子液体类
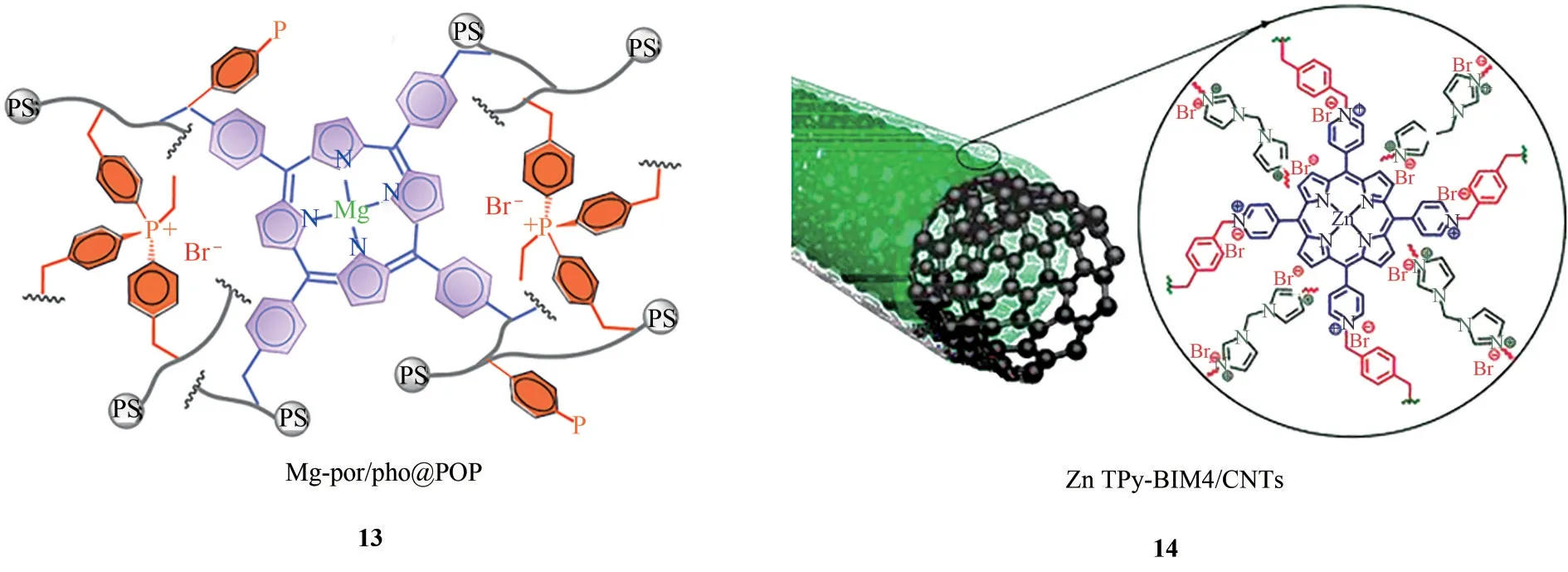
图14 Mg-por/php@POP和ZnTPy-BIM4/CNTs的结构示意图
2016 年,Leng 等[72]首先利用4,4'-联吡啶与1,2,4,5-四溴甲基苯发生季铵化反应,得到富含吡啶离子液体和溴甲基的多孔聚合物TBB-Bpy,再利用溴甲基对Salen Co 单体中叔氨基的季铵化反应,成功制备得到多功能聚合物TBB-Bpy@Salen Co。遗憾的是,该聚合物具有非常小的比表面积(<3m2/g)。尽管如此,TBB-Bpy@Salen Co中同时包含丰富的吡啶离子液体、季铵盐和Salen Co活性基团,依然可作为协同催化剂用于CO2与环氧化物合成环状碳酸酯的反应中。当不加入任何溶剂和助剂,在60℃和1MPa 条件下反应6h,可获得99.2%的PO转化率和100%的PC选择性。
严格来说,上述致密的多功能聚合物TBBBpy@Salen Co并不能被称为多孔材料。因此,基于金属Salen 配合物的POPs 材料是Cao 课题组[73]于2016 年首次合成。该课题组以咪唑基三嗪化合物和氯甲基功能化的Salen Al为原料,溶剂热条件下合成了同时包含咪唑离子液体和Salen Al单元的多孔有机聚合物Al-CPOP(图15,15),其比表面积达到136m2/g,孔径分布集中于2.5nm 左右,为典型的介孔材料。该作者将其成功应用于催化CO2与环氧化物加成生成环状碳酸酯的反应,但反应温度(120℃)依然较高,产物选择性也待进一步提高。不久后,Ji 和Luo 课题组[74]开发了乙烯基咪唑离子液体功能化的金属Salen配合物,并将其与DVB通过自由基共聚反应,获得了具有高比表面积的微介孔聚合物DVB@ISA (SBET=590m2/g) 和DVB@ISZ(图15,16)。DVB@ISA 在催化无溶剂、无助剂条件下的CO2环加成反应时,表现出良好的催化活性,60℃和1MPa 下反应24h,可获得99%的PC 收率。相比于Cao课题组的Al-CPOP催化体系,该体系具有更优异的催化活性和选择性。该作者认为,DVB@ISA 具有更高的孔隙率,且其丰富的多级孔结构有利于催化活性位点的暴露以及底物和产物的传质,进而表现更高的催化活性。另外,催化剂框架中丰富的苯环结构使得其表现疏水特性,进而在催化反应过程中表现出更高的化学选择性和稳定性。
最近,Li等[75]利用Friedel-Crafts 烷基化反应与季铵化反应成功制备得到多孔金属Salen 基超交联离子聚合物。同样,在CO2环加成反应中,由于框架中金属中心和卤素阴离子的协同效应,该类双功能多孔聚合物也表现出良好的催化剂性能和良好的孔隙率,比表面积可达631m2/g。相对于其他合成策略,该方法在改进离子聚合物的孔结构以及增大比表面积方面表现出一定的优势,同时原料易得、合成过程也简单,因此为获得高比表面积和良好孔隙率的多孔离子聚合物提供了一条可行的路径。
3.3 其他金属配合物/离子液体类
2016 年,Ding 等[76]首先通过自由基共聚反应,将乙烯基功能化的三苯基膦和乙烯基功能化的咪唑离子液体聚合得到PPh3-ILBr@POPs。然后采用浸渍法,将ZnX2(X=Cl,Br,I)负载于上述多孔聚合物中,利用其框架中丰富的磷位点对Zn2+的配位稳定作用,获得了PPh3-ILBr-ZnX2@POPs(X=Cl,Br,I)。基于聚合物中同时含有Lewis 酸性的Zn2+和亲核试剂Br-,其可作为高效催化剂协同催化CO2环加成反应生成环状碳酸酯。同时,该作者利用在线红外对反应进程进行跟踪,为详细考察各反应条件对催化性能的影响提供了更直观、细致的辅助作用。随后,该课题组[77]利用相同的合成策略,将咪唑离子液体替换为季鏻盐,并获得了一系列多孔离子聚合物材料P+Br--PPh3@POPs。该作者将所得离子型聚合物应用于CO2与环氧化物的环加成反应中。通过调节季鏻盐和三苯基膦(PPh3)的比例,优化催化活性,得出在相同反应条件下,1P+Br-&1PPh3@POPs 表现出最优的催化性能。该作者将此解释为大量的PPh3引入聚合物中,导致季鏻盐活性组分被掩埋。此外,考虑到PPh3能够与ZnBr2等金属配位,从而引入Lewis 酸活性位点。大量的PPh3的引入使得配位后的Zn2+与季鏻盐相距太远难以产生协同效应。利用浸渍法将不同的金属卤盐(IrCl3、RuCl3、RhCl3、YCl3、CoCl2、ZnCl2、ZnI2和ZnBr2)负载于所得双功能多孔聚合物,筛选出最优催化剂为1P+Br-&ZnBr2-1PPh3@POPs,不加入溶剂和助剂时,在120℃和3MPa 下,初始TOF 值可达3985h-1。

图15 Al-CPOP、DVB@ISA和DVB@ISZ的结构示意图
3.4 氢键供体/离子液体类
Dyson 等[78]在2017 年报道了一种双乙烯基苯基咪唑离子液体,通过AIBN 引发自由基聚合反应,形成有机多孔材料[pna/b(n=1~6)],结构如图16所示。聚合物p1a/b、p2a/b 和p3a/b 中只含有离子液体催化活性组分,p4a/b、p5a/b 和p6a/b 中除了包含离子液体基团外,还分别引入了一元醇羟基、邻二醇基团以及羧基。这些引入的基团可作为HBD位点与环氧化物中的氧原子形成氢键,从而活化环氧化物。该作者将所得的聚合物应用于CO2环加成反应,实验结果也表明,具有双重活性组分的催化剂表现出更高的催化活性。作为最优催化剂p5b,在2.5MPa 和130℃条件下,无需助催化剂,15h 可实现对多种环氧化物90%以上的转化率。催化剂重复使用4次后,其活性和选择性几乎无降低。
同年,Yang课题组[79]首先利用自由基共聚反应获得了一系列含有醇羟基的咪唑离子液体聚合物PxVyOHzR。该作者通过调控单体之间的比例,发现当DVB 比例超过0.70 时所获得的P0.70V0.15OH0.15R1为介孔材料(SBET=365m2/g),而其比例小于0.25 时所获得的P0.25V0.15OH0.60R1和P0.25V0.15OH0.60R2则为油状物。对此,该作者解释为DVB 的用量减少,使得交联程度大大降低而难以获得固体材料。以上聚合物在CO2环加成反应中均表现出良好的催化性能。在2MPa 和140℃条件下反应5h,多相催化剂P0.25V0.15OH0.60R1和均相催化剂P0.25V0.15OH0.60R2分别可获得84%和95%的PC收率,相应的初始TOF 值为39h-1和71h-1。尽管如此,考虑到反应过程中P0.25V0.15OH0.60R2溶于反应体系,使得其难以回收和循环使用。该作者进一步设计了甲基丙烯酸酯基功能化的SBA-15作为单体,将其与乙烯基咪唑离子液体和对乙烯基苯乙醇聚合后,得到了SBAV0.15OH0.60R237 催化剂,其比表面积为259m2/g。在相同条件下,催化剂P0.25V0.15OH0.60R2可获得与SBAV0.15OH0.60R237 相同的PC 收率。 然而, SBAV0.15OH0.60R237 的初始TOF 值有了很大提高(188h-1vs.71h-1)。该作者将此解释为:在SBA-V0.15OH0.60R237体系中,反应主要发生在密闭的介孔中,而密闭的纳米孔道有利于提高离子液体中阴离子的活化能力。

图16 pna/b(n=1~6)的结构示意图
2017 年,Zhang 等[80]利用微流控技术,通过自由基共聚法和后合成策略,可控制备了粒径分别为67nm、271nm 和375nm 的球状多孔聚合物NPILs-BPA。该聚合物框架中同时包含季鏻盐和羧基,可作为双功能催化剂用于协同催化CO2与环氧化物生成环状碳酸酯的反应中。实验结果表明,在150℃和2MPa 条件下,催化剂NPILs-BPA 表现出良好的催化性能和重复使用性。该作者利用原位红外手段实时监控对照反应的进程,直观地证实了该反应是在季鏻盐和羧基的协同催化下进行的。此外,该作者开发的新颖而连续的微流控技术也为今后纳米催化剂的可控制备提供了一种重要合成途径。随后,Wang 课题组[81]利用自由基聚合以及后续环氧基开环方法获得了富含邻二羟基的多孔离子液体聚合物PDGBr-2OH。该类双功能催化剂可在0.1MPa和90℃条件下高效催化该环加成反应。基于HBD/ILs 协同活化机制,该作者利用量子化学计算对邻位双羟基协同作用于环氧化物的过程进行详细分析,也对该催化体系中的反应机理进行推测,对今后类似催化剂催化的反应机理具有一定的参考价值。
不久后,Ji 和Luo 等[82]利用溶剂热条件下的酚醛树脂聚合反应合成了一系列可用于协同催化CO2环加成反应的多相催化剂FIP-Im(图17)。值得注意的是,基于该反应的前合成策略(pre-synthetic method),有效避免了自由基聚合反应过程中酚羟基的阻聚作用,从而获得了富含酚羟基和咪唑离子液体的聚合物。遗憾的是,由于大量离子液体的引入,使得该聚合物比表面积较小。尽管如此,由于框架中含有大量的极性离子液体基团和氮、氧原子,该聚合物表现出良好的CO2亲和性和选择性。在0℃和1bar 条件下,FIP-Im 的CO2吸附量为1.45mmol/g,CO2/N2吸附选择性为36∶1。FIP-Im在催化上述反应时依然表现出优异的催化性能:1MPa 和80℃条件下,无需加入任何溶剂和助剂,反应10h可获得99%的PC收率。此外,基于FIP-Im框架中的酚羟基具有易修饰性,该作者采用后合成法,通过Williamson成醚反应对其进行功能化。该作者基于文献调研,以苯硅烷为还原剂时,季铵盐能够高效催化CO2与亚胺反应生成N-甲酰胺类化合物[83-85]。因此,将季铵盐锚定在FIP-Im 框架中,并将所得高密度离子活性中心的催化剂FIPIm@QA(图17)应用于上述N-甲酰化反应中,在35℃和1MPa 条件下,不加入任何助剂和溶剂时,可将N-甲基苯胺完全转化为N-甲基甲酰苯胺。值得注意的是,FIP-Im 和FIP-Im@QA 也可以将模拟废气(15%CO2+85%N2,体积分数)顺利转化为相应模型反应的产物。该工作中,作者耦合了“前合成”与“后合成”策略用于设计功能导向型的功能聚合物催化剂,也实现了绿色、温和条件下CO2原价插入反应与CO2还原反应生成高附加值的化合物。
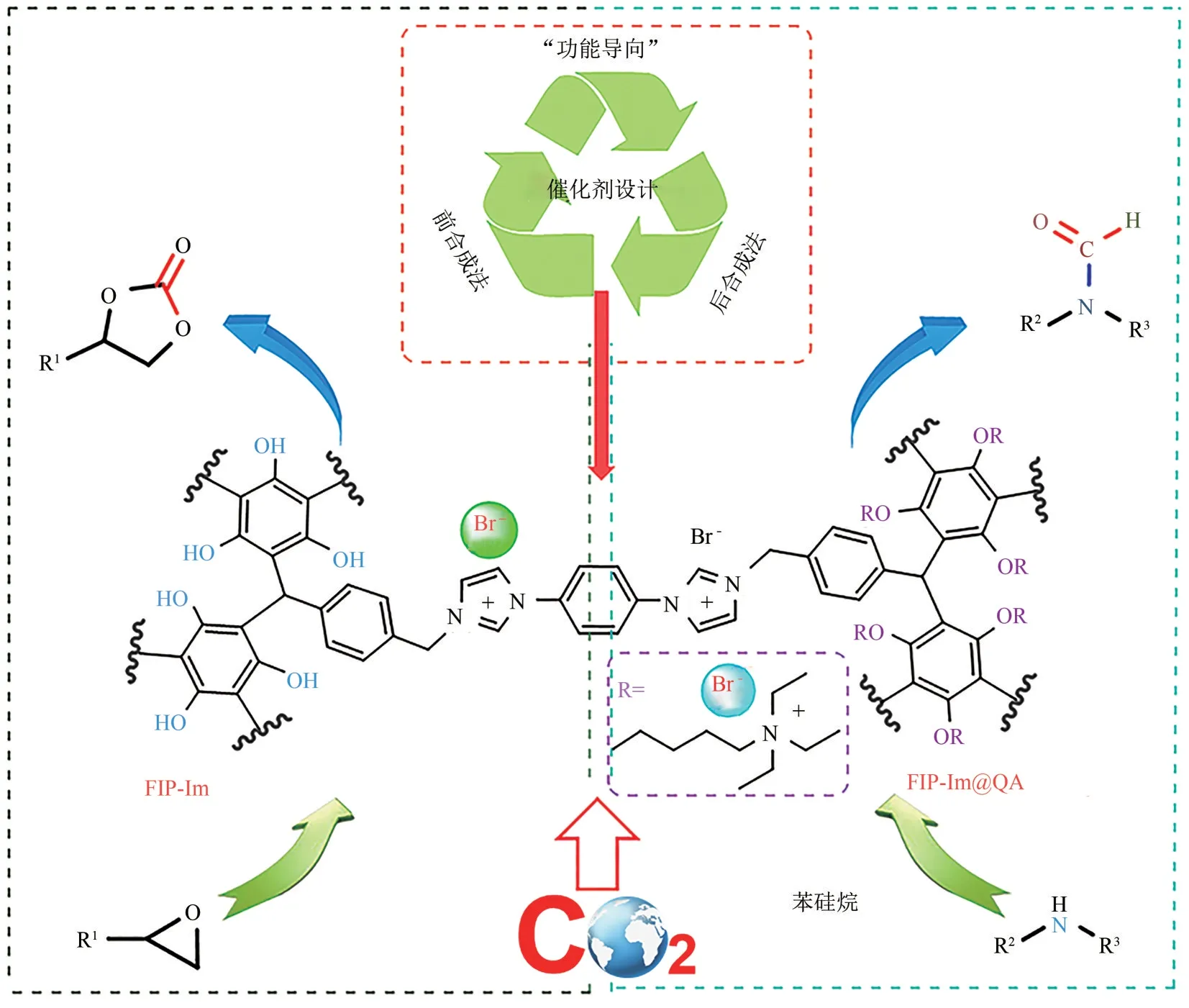
图17 FIP-Im和FIP-Im@QA分别在CO2环加成反应和N-甲酰化反应中的应用[82]
最近,Sun 和Xiao 等[86]采用从头合成策略制备了一系列具有较高比表面积的季鏻型多孔聚合物,结构如图18所示。在PPS-oOH-Bn、PPS-mOH-Bn和PPS-pOH-Bn框架中,酚羟基分别位于与磷原子相连亚甲基的邻位、间位和对位。该作者以正丁基缩水甘油醚与CO2的环加成反应为模型,研究了酚羟基和溴离子这两种功能位点的相对位置对催化性能的影响,阐明了酚羟基的重要作用以及双功能催化剂中活性位点位于合适位置而产生的良好协同效应。进一步,该作者通过密度泛函理论计算方法获得了所推测机理中基态到过渡态的活化能(PPSpOH-Bn>PPS-oOH-Bn>PPS-mOH-Bn),表明含间位酚羟基的催化剂表现出最高活性,这与实验结果一致。该作者通过上述研究,提出可通过设计和调节催化剂中活性组分之间的相对位置来放大它们之间的协同效应,进而优化催化材料。毫无疑问,该工作所阐述的结构性能关系对其他协同催化剂的结构设计与性能优化也具有借鉴意义。
3.5 其他含二元活性组分的POPs催化剂
2016 年,Wang 等[87]精心设计了氨基功能化的乙烯基咪唑离子液体单体,通过自由基聚合反应和离子交换获得了阴离子为氢氧根的咪唑离子液体聚合物PAD-3(图19)。该聚合物框架中富含氨基,比表面积为156m2/g,孔径分布范围为3.0~3.9nm。在应用于CO2与环氧化物环加成反应中时,该聚合物中的氢氧根和氨基作为有机碱可高效活化CO2,从而使得该反应能够顺利进行。在70℃和0.1MPa下反应24h,即可将环氧氯丙烷全部转化为相应的环状碳酸酯。值得注意的是,大位阻环氧环己烷往往需要在非常苛刻条件下才能顺利反应。然而,利用该聚合物在70℃和1MPa 条件下反应15h,即能够获得94%的产物收率,这为开发能够高效催化转化大位阻环氧化物与CO2发生环加成反应的多相催化剂提供了指导。
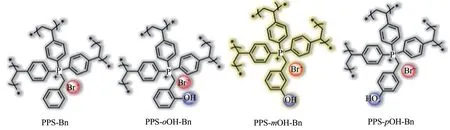
图18 季鏻型多孔聚合物的结构
图19 PAD-3的合成路线图及其在CO2与环状碳酸酯的环加成反应中的应用[87]
同年,Liu 等[88]以均苯三酚和4,4'-二氨基联苯为原料,通过在水溶液中发生重氮偶联反应,得到了一种富含酚羟基的多孔聚合物HAzo-POP-1,该方法具有绿色、简捷和高效的优势。随后,利用酚羟基与金属盐的配位作用,制备得到一系列同时包含酚羟基和金属离子活性位点的多功能催化剂。以上配位负载过程也被XPS表征所证实。该作者将该类催化剂应用于以TBAB 为助催化剂的CO2/环氧化环加成反应中。其中,Zn/HAzo-POP-1 在100℃和0.1MPa 条件下反应0.5h 可获得90%的PC 收率,TOF 值高达2888h-1。值得注意的是,在常温常压条件下,将反应时间延长至48h,所有PO 全部转化为PC。近期,Dai 课题组[89]结合无溶剂自组装和嵌段共聚物模板法也获得含丰富酚羟基的氮掺杂介孔材料N-OMPs。负载Zn2+和Co2+后,获得的多功能催化剂也成功应用于CO2环加成反应中,并表现出良好的催化性能和稳定性。
最近,Chen等[90]采用含酚羟基的苯甲醛类化合物与吡咯反应,通过原位形成卟啉配体及后上金属的方法,构建了富含酚羟基和锌卟啉的多孔聚合物P-POF-Zn。该聚合比表面积可达639m2/g,孔径集中分布于0.5~1.3nm。在以[CPeDMAPy]Br为助催化剂,100℃和1.5MPa CO2条件下反应12h 可获得99%的PC 收率。该作者还考察了0~40%(摩尔分数)([H2O]/[PO])的水对该反应的影响。结果表明,在0~15%(摩尔分数)范围内,随着水量的增加,对该反应有一定的促进作用,并且对其产物选择性没有明显影响。而进一步增加水用量时,虽然其催化活性基本无变化,但产物选择性则有了一定下降。该作者认为在较低的水用量范围内,水与助催化剂[CPeDMAPy]Br 可协调作用而加速PO 开环,然而继续增加水用量后,PO 的水解开环反应使得产物选择性下降。
近期,Wu 等[91]制备了一种高度有序的咪唑功能化的介孔酚醛树脂(IPMPs)(图20)。制备过程中发现,IPMP 介孔结构的顺序在很大程度上依赖于咪唑基前体。3-(咪唑-1-基)苯酚与未占据的邻位和对位被发现是最佳前体,生成高度有序的交联聚合物(3-IPMP),且具有高的比表面积、低密度、均匀的中孔和可控制的咪唑负载量。框架中丰富的咪唑和酚羟基可协同活化CO2,在添加碘化钾后,能够实现环状碳酸酯的高效和高选择性合成。基于以上结果,该作者进一步利用碘乙烷将3-IPMP 框架中的咪唑离子化,获得离子型聚合物3-IPMP-EtI。如此一来,在3-IPMP-EtI应用于CO2环加成反应中时,有效避免了碘化钾的添加。Ahn等[92]以苯并咪唑离子液体、三苯基苯和甲醛二甲基缩醛为原料,采用Friedel-Crafts烷基化反应合成了一种新型纳米孔氮杂环卡宾型交联离子聚合物(NHC-CAP-1),并利用溴化锌对其进行了进一步金属化。所得的双功能催化剂的比表面积高达1040m2/g,在273K和1bar条件下的CO2吸附量也达到188.2mg/g。该合成方法简单高效,且在催化CO2环加成反应中展现出良好的工业应用前景。
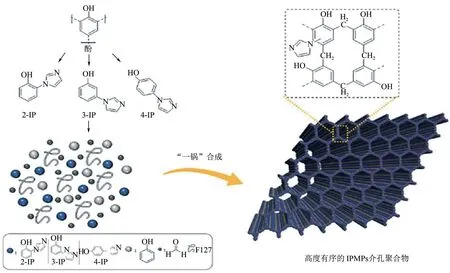
图20 高度有序且可调控的咪唑基介孔聚合物IPMPs的合成[91]
4 结语与展望
基于全球面临环境日趋恶化和资源严重匮乏的现状,合理有效利用现有资源,发展绿色化学以顺应可持续发展战略已经势在必行。基于CO2的固有属性,将其捕获并转化为高附加值的化学品,是实现人工碳资源循环的重要途径。CO2与环氧化物制备环状碳酸酯的反应是CO2资源化利用与存储中研究与应用最为广泛的反应之一。基于该反应机理的调研,酸碱协同催化机制具有高效、高选择性以及条件温和的特点,甚至能够实现常温常压下CO2的活化及转化。尤其,基于新兴的多孔有机聚合物(POPs)材料的多相催化体系取得重要研究进展。从该类催化材料的发展过程来看,虽然起步较晚,但是近年来实现了从一元活性组分的POPs 体系至二元乃至多元活性组分的POPs 的跨越式发展。POPs 在催化CO2与环氧化物环加成制备环状碳酸酯的反应中展现出优异的催化活性和选择性,并且可实现催化剂简捷回收和稳定重复使用。这些成果不仅推动了多孔有机材料的发展,也促进了CO2制备高附加值化学品反应的工业化应用进程。
POPs 的特性之一是可通过多样的化学反应和先进合成策略制备得到;另一特性是在原子层次上的单体设计具有相当大的灵活性,能够合成具有不同结构和组成的聚合单体。这两种特性有助于优化该类材料的孔径以及使得其比表面积最大化。更重要的是,具有不同结构和功能的多孔聚合物能够适用于特定的领域。目前,文献报道的大多为电中性的多孔有机聚合物。离子组分的引入不仅能够增加多孔有机聚合物种类的多样性,也将进一步拓宽其应用范畴。多孔离子聚合物与前者类似,可通过设计中性单体调节其比表面积。另外,还可以通过离子交换等方式,从原子或分子水平调控其结构和功能特征。然而,由于电荷相互作用以及分子间发生的堆积效应,该类带电荷的聚合物通常具有较低的比表面积和较小的孔容,这也成为制约其广泛应用的一个重要因素。尤其在用作催化剂时,低的比表面积和孔隙度不利于活性位点暴露和分子扩散。因此,设计具有一定刚性和扭曲特性的功能单体,并选择合适的聚合反应与策略,是维持该类多孔离子聚合物较高比表面积和良好孔隙度的重要前提。
基于POPs 在CO2吸附、选择性吸附方面的优势以及聚合反应的多样性,开发功能型有机多孔聚合物,能够有效地将CO2捕集和后续转化耦合。在温和条件下实现CO2高效捕集并转化为高附加值的化学品,需注意以下几个方面:①聚合反应简单、高效和环境友好;②引入具有活化底物和/或CO2的高活性基团;③引入极性基团或者杂原子,加强框架对CO2分子的相互作用;④引入多级孔结构、微孔结构改善CO2富集能力,介孔或大孔利于底物与产物传质;⑤提高活性位点密度,增加其与反应物碰撞概率,强化多个活性位点协同作用。此外,基于CO2活化方式的多样性,有导向性地引入功能基团,将有利于拓展POPs 材料在CO2参与的其他有机反应中的应用。最后,随着量子化学计算的不断发展,将其应用于催化反应历程的推测并指导催化剂的合成也是今后重要的发展方向。
