nZVI/PMS去除污染物机制及应用过程中控制参数对去除效果的影响
2021-07-25关英红张天禹孙维敬
关英红,张天禹,孙维敬,丁 红
(1.东北农业大学水利与土木工程学院,哈尔滨 150030;2.黑龙江水利科学研究院,哈尔滨 150050)
阿特拉津(ATZ),又名莠去津,1952年由瑞士Geigy公司研发,是一种高效除草剂[1]。研究表明ATZ是一种毒性内分泌干扰物[2],对人和哺乳类生物有致癌作用[3-4]。土壤中ATZ易通过降水作用进入地表水和地下水[5]。水中残留ATZ危害人类饮用水安全。ATZ结构稳定难以自然降解,常规氧化降解工艺效果不佳,研究表明基于过氧化氢、过硫酸盐的高级氧化技术均可实现对水中ATZ的高效降解[6-7]。
铁是地球上最常见过渡金属元素,其化学性质活泼,价格低廉,广泛用于环境修复[8]。Fe2+活化过硫酸盐是降解有机污染物有效方式,然而Fe2+在中性和碱性条件下易沉淀,同时Fe2+浓度难以控制,过量Fe2+会消耗·SO4-,影响污染物降解效果,限制Fe2+均相活化过硫酸盐在工程实践中应用[9]。零价铁催化剂在非均相活化过硫酸盐氧化体系中被广泛使用。零价铁催化剂具有磁性,方便回收利用,节约成本[10]。
本文通过液相还原法制备纳米尺寸零价铁(nZVI)并对其表征,分析nZVI与PMS反应前后结构和元素价态变化;以ATZ为目标污染物,甲醇(MA)和叔丁醇(TBA)为自由基捕获剂,结合反应过程中PMS浓度变化分析nZVI/PMS体系降解ATZ反应机理;研究零价铁在应用过程中控制参数(颗粒尺寸和循环使用次数)对ATZ去除效果的影响。为nZVI/PMS体系降解自然水体中ATZ提供理论依据。
1 试剂与方法
1.1 试验试剂与仪器
1.1.1 试验试剂
主要试剂如下:磷酸、阿特拉津、氢氧化钾、单过硫酸盐化合物均购自西格玛奥德里奇(上海)贸易有限公司;邻二氮菲、乙酸、乙酸钠、硼氢化钠、硫酸、邻苯二甲酸氢钾、盐酸羟胺、碘化钾均购自上海国药集团化学试剂有限公司;还原铁粉规格为直径150μm,购自阿拉丁试剂(上海)有限公司;七水合硫酸亚铁、无水乙醇、叔丁醇、钼酸铵购自天津天力化学制剂有限公司;甲醇购自上海赛默飞世尔科技有限公司。其中,甲醇为色谱纯;其余试剂均为分析纯。所有试验溶液若无特殊说明均采用Milli-Q超纯水配置。
1.1.2 试验仪器
主要仪器如下:精密酸度计(PHS-3C);恒温水浴振荡器(SHZ-B);紫外分光光度计(T6,MC China);电子分析天平(SQP);高效液相色谱仪(Waters Acquity UPLC H-Class);真空干燥箱(DZ-1BCIV);机械搅拌器(JJ-1B);场发射扫描电子显微镜(FEI Quanta25);X射线光电子能谱分析仪(EscaLab 250Xi)。
1.2 试验方法
1.2.1 nZVI制备
采用液相还原法制备nZVI[11]。称取16.68 g七水合硫酸亚铁溶解于120 mL水中,将制备好硫酸亚铁溶液加入四口烧瓶,加入280 mL无水乙醇,在氮气保护下机械搅拌10 min(转速200 r·min-1),随后用恒压分液漏斗向四口烧瓶中缓慢滴加100 mL硼氢化钠溶液(c=1.59 mol·L-1)。在氮气保护下机械搅拌30 min形成固液混合物(转速200 r·min-1)。通过强磁分离得到黑色固体,经无水乙醇和无氧超纯水分别洗涤3次后放入真空干燥箱,在100℃条件下真空干燥10 h制备nZVI固体,nZVI固体研磨成粉末状后真空保存备用。
1.2.2 nZVI表征
采用场发射扫描电子显微镜(FEI Quanta25)分别分析nZVI及其活化PMS后材料表面微观样貌,仪器工作电压20 kV,放大倍数80 000倍;分辨率3.73μm。
采用X射线光电子能谱(EscaLab 250Xi)表征nZVI及其活化PMS后主要元素组成和价态变化,该仪器采用电子能量为1 486.6 eV的Al Kα单色光源(Mono AlKα),束斑尺寸为400μm,扫描模式为CAE,扫描5次。
1.2.3 nZVI/PMS降解ATZ反应机理探究试验
共6组试验,将装有反应液的250 mL三角瓶置于转速200 r·min-1,温度25℃恒温水浴振荡器中,试验开始前预热15 min。将150 mL ATZ/PMS反应液(cATZ=2.1μmol·L-1、cPMS=30μmol·L-1)pH调至3,其中4组分别加入3 mL MA溶液、15 mL TBA溶液、3 mL TBA溶液、15 mL MA溶液;1组不添加MA、TBA溶液;1组不添加MA、TBA溶液且反应液中不添加PMS。6组反应液分别投入0.03 g nZVI粉末开始计时,设定反应时间点时注射器取样1.5 mL,过直径0.45μm滤膜加入至液相小瓶中用于ATZ浓度分析。所有液相小瓶中提前加入50 μL甲醇和20μL盐酸羟胺溶液(c=5 mol·L-1)作为反应终止剂。其中MA溶液配置方法为称取0.32 g MA用水定容至100 mL;叔丁醇溶液配置方法为称取0.74 g TBA用水定容至100 mL。所有试验初始pH均由0.184 mol·L-1硫酸调节,溶液pH均使用上海雷磁精密酸度计(PHS-3C)测定。
PMS浓度变化试验:到设定时间反应点取3 mL样品过直径0.45μm滤膜,分析PMS浓度。
溶液中总铁以及二价铁含量试验:反应10 min后分别取5 mL样品过直径0.45μm滤膜,分析总铁及二价铁含量。总铁与二价铁分析方法使用邻二氮菲吸收光谱法[12]。取5 mL样品加入比色管中,向比色管中加入0.2 mL 10%(M/V)盐酸羟胺溶液摇匀静置2 min后,依次加入0.4 mL 0.15%(M/V)邻二氮菲溶液、1 mL pH 4.6乙酸乙酸钠缓冲溶液,摇匀定容至10 mL制成待测样。静置10 min后用移液枪取约2 mL待测样加入10 mm光程石英比色皿中,在波长λ=508 nm条件下使用紫外分光光度计分析。
1.2.4 不同尺寸零价铁对ATZ降解效果影响试验
试验方法与1.2.3相同,不添加TBA、MA溶液。一组投入0.03 g nZVI粉末,对照组投入0.03 g微米零价铁粉(mZVI)开始计时,到设定反应时间点取样,分析ATZ和PMS浓度。ATZ浓度分析方法使用液相色谱(Waters Acquity UPLC H-Class)分析。HPLC工作条件如下:色谱柱为Waters Acquity UPLC®BEH C18 column(2.1 mm×100 mm,1.7μm颗粒),检测器为PDA检测器,流动相为甲醇-磷酸水溶液(60∶40,V/V),其中磷酸水溶液浓度为6 mmol·L-1;流速0.1 mL·min-1,进样量10μL,色谱柱温度25℃,紫外检测波长226 nm。在此检测条件下,ATZ出峰时间为4 min。
PMS浓度分析方法使用紫外分光光度计法[13]。取1.5 mL Solution A、1.5 mL Solution B、3 mL样品在比色管中均匀混合静置15 min制成待测液,用移液枪取2 mL待测液加入10 mm光程石英比色皿中,在波长λ=351 nm条件下使用紫外分光光度计(T6,MC China)分析。其中Solution A配置方法为邻苯二甲酸氢钾10 g溶于500 mL水;Solution B配置方法为四水合钼酸铵0.1 g、碘化钾33 g、氢氧化钾1 g溶于500 mL水。
1.2.5 nZVI/PMS降解ATZ体系nZVI重复利用效果探究试验
试验方法与1.2.3相同,不添加TBA、MA溶液。反应10 min后将溶液中固体强磁分离,超纯水洗涤3次,以相同条件反应,到设定反应时间点取样,分析ATZ浓度。
无特殊说明上述所有试验作两次重复,结果取平均值。
2 结果与分析
2.1 nZVI表征结果分析
2.1.1 形貌和结构分析
通过场发射扫描电子显微镜表征nZVI及其活化PMS后表面形貌和结构。图1a表明nZVI单个颗粒为球形结构;nZVI颗粒由于磁性和表面张力作用呈团聚现象。图1b表明nZVI与PMS反应后,纳米材料颗粒出现无规则片状结构。图1c表明nZVI颗粒表面较光滑,具有较小颗粒尺寸,推测直径为200~300 nm。

图1 催化剂SEM图像Fig.1 SEMimages of catalyst
2.1.2 元素组成价态分析
通过X射线光电子能谱表征nZVI及其活化PMS后主要元素组成和价态变化。
图2 a显示nZVI的XPS扫描全谱。图2a表明,nZVI在712和725 eV处出现两个峰,分别对应Fe 2p3/2和Fe 2p1/2结合能;检测到C 1s信号峰和较为强烈的O 1s信号峰。nZVI与PMS反应前后Fe 2p窄谱图如图2b、c所示,nZVI与PMS反应前检测到明显Fe0特征峰(706.7 eV),反应后检测不到Fe0特征峰。

图2 催化剂XPS光谱图Fig.2 XPS spectra of catalyst
2.2 nZVI/PMS降解ATZ反应机理探究
Anipsitakis等研究表明,MA与·SO4-和·OH反应速率相差小,可作为·SO4-和·OH淬灭剂;TBA与·OH反应速率是其与·SO4-反应速率近1 000倍,可作为·OH淬灭剂[14]。为探究nZVI/PMS体系降解ATZ中自由基种类,向体系中分别加入MA和TBA开展自由基淬灭试验。结果如图3所示,在nZVI投量0.2 g·L-1、PMS初始浓度30μmol·L-1、ATZ初始浓度2.1μmol·L-1、初始pH=3条件下,反应10 min,未添加自由基淬灭剂时ATZ降解率为100%;TBA、MA浓度为2 mmol·L-1时ATZ降解率分别为68.1%和57.6%;TBA、MA浓度为10 mmol·L-1时ATZ降解率分别为27.1%和23.9%;仅投加nZVI不添加PMS时ATZ降解率为0。

图3 不同浓度TBA、MA对ATZ降解效果影响Fig.3 Effect of different concentrations of TBA and MA on degradation of ATZ
图4 显示在nZVI投量0.2 g·L-1、PMS初始浓度30μmol·L-1、ATZ初始浓度2.1μmol·L-1、初始pH=3条件下反应10 min,PMS浓度变化。随反应进行,反应液中PMS浓度不断降低。通过邻二氮菲吸收光谱法测得该体系下反应10 min后溶液中总铁浓度为1.9×10-5μmol·L-1,Fe2+浓度为9.1×10-6μmol·L-1。

图4 nZVI/PMS降解ATZ体系PMS浓度变化Fig.4 Change of PMS concentration in nZVI/PMS degradation ATZ system
2.3 不同尺寸零价铁对ATZ降解效果影响
固体催化剂尺寸影响其比表面积,可能影响其催化性能。为探究固体粒径对零价铁活化PMS降解ATZ效果影响,选取平均颗粒直径为150μm(100目)mZVI作对照。图5表明,粒径尺寸对ATZ降解效果存在影响。在催化剂投量0.2 g·L-1、PMS初始浓度30μmol·L-1、ATZ初始浓度2.1μmol·L-1、初始pH=3条件下,采用mZVI作催化剂时,10 min反应时间ATZ降解率为89.98%;采用nZVI作催化剂时,10 min反应时间ATZ降解率为100%。图6表明,nZVI作催化剂时,反应10 min后PMS浓度为初始浓度32.9%;mZVI作催化剂时,反应10 min后PMS浓度为初始浓度38.9%。
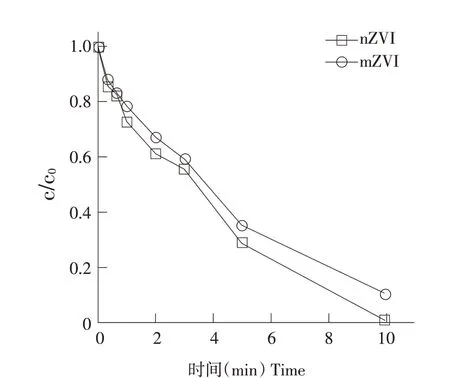
图5 不同尺寸零价铁对ATZ降解效果影响Fig.5 Effect of zero valent iron with different sizes on degradation of ATZ

图6 不同尺寸零价铁对PMS活化能力影响Fig.6 Effect of zero valent iron with different sizes on activation ability of PMS
2.4 nZVI/PMS降解ATZ体系nZVI重复利用效果探究
在nZVI/PMS体系降解ATZ试验中,nZVI仅有一部分参与到催化反应过程,反应结束后溶液中残留一部分固体nZVI。为探究该部分nZVI可利用性及催化活性,对nZVI作重复使用试验。为保证每次反应均在催化剂投量0.2 g·L-1、PMS初始浓度30μmol·L-1、ATZ初始浓度2.1μmol·L-1、初始pH=3条件下完成,需计算每次反应nZVI损失率。具体方法为使用盐酸溶液溶解反应10 min后经洗涤的剩余nZVI,通过邻二氮菲吸收光谱法测定总铁浓度计算nZVI损失率。再通过计算所需药剂量确保每次重复使用试验条件相同。经计算上述试验条件下每次nZVI损失率为11%。如图7所示,第1次反应10 min时,ATZ降解率为100%;第2次反应10 min时ATZ降解率为85.2%;第3次反应10 min时ATZ降解率为83.9%。

图7 nZVI/PMS降解ATZ体系nZVI重复利用效果Fig.7 Effect of nZVI/PMS on degradation and reuse of nZVI in ATZ system
3 讨 论
3.1 nZVI表征
由图1可知,nZVI颗粒呈球形,由于磁性和表面张力作用呈团聚现象,与胡书红报道nZVI具有核壳状状结构一致[15];与PMS反应后呈现无规则片状结构,推测可能原因为反应过程中纳米材料表面被氧化,腐蚀的碎片堆积在纳米颗粒表面形成片状结构。
由图2可知,nZVI在结合能为706.7 eV处存在明显峰,该处对应Fe0特征峰。nZVI在712和725 eV处出现两个峰,分别对应Fe 2p3/2和Fe 2p1/2的结合能。对其拟合发现,nZVI表面同时存在Fe2+和Fe3+(见图2b)。同时nZVI表面也检测到较强烈O 1s信号峰,说明纳米颗粒表面被氧化。此外,nZVI表面检测到C 1s信号峰,推测可能原因为nZVI接触到空气中含炭物质[16]。由图2c拟合曲线结果可知,零价铁表面存在Fe2+和Fe3+。但反应后对应结合能为706.7 eV的Fe0特征峰消失,推测其可能原因为nZVI颗粒表面在与PMS反应过程中进一步氧化,表面可能完全被二价铁和三价铁氧化物所覆盖,氧化层变厚。
3.2 nZVI/PMS降解ATZ反应机理探究
由图3~4可知,nZVI/PMS体系对ATZ具有较好降解效果。在nZVI投量0.2 g·L-1、PMS初始浓度30μmol·L-1、ATZ初始浓度2.1μmol·L-1、初始pH=3条件下反应10 min,未添加自由基淬灭剂时ATZ降解率为100%。且单独的nZVI对ATZ无明显吸附去除作用。TBA、MA浓度在2 mmol·L-1时对nZVI/PMS体系下ATZ降解有明显抑制作用且同等浓度下MA抑制作用强于TBA,当TBA、MA浓度增大10 mmol·L-1时,抑制作用更明显;说明体系中产生的·SO4-和·OH均是降解ATZ的主要氧化活性物种[17-18]。nZVI与PMS在水中溶解生成Fe2+(方程1),Fe2+与HSO5-反应生成·SO4-或·OH(方程2和3),·SO4-可与H2O反应生成·OH(方程4~5)共同氧化ATZ实现降解[19]。

3.3 不同尺寸零价铁对ATZ降解效果影响
图5 、6表明,nZVI催化PMS降解ATZ效率高于mZVI体系,说明零价铁尺寸越小,活化PMS降解ATZ效果越好。
结合SEM表征结果(见图1),推测原因为nZVI拥有独特核壳结构,具有较大比表面积。Li等使用不同尺寸零价铁(1 mm零价铁、150μm零价铁、50 nm零价铁)活化PS降解酸性橙7时得出类似结论:零价铁尺寸越小,对PS活化效果越出色[20]。取nZVI颗粒直径为250 nm,mZVI颗粒直径为150μm,假设二者结构为均匀球体且密度相同,经计算等质量两种催化剂比表面积比S纳米零价铁∶S微米零价铁=100∶1.67,纳米零价铁与微米零价铁活化PMS降解ATZ假一级常数比k纳米零价铁∶k微米零价铁=1.03∶1(R2纳米零价铁=0.97;R2微米零价铁=0.99),可看出两种试验条件下假一级速率常数比值远小于比表面积比值,说明ATZ降解效果并非与零价铁颗粒比表面积呈正比增加,推测原因为降低nZVI颗粒尺寸,虽然增加比表面积,增大Fe0/PMS体系产生活性自由基速率,但同时与污染物ATZ竞争捕获自由基的铁含量也增加。
3.4 nZVI/PMS降解ATZ体系nZVI重复利用效果
图7 表明,尽管ATZ降解率随nZVI使用次数增加而降低,但在第3次使用时nZVI/PMS体系对ATZ仍有较好降解效果。结合SEM图像和XPS谱图分析,认为nZVI重复使用时对ATZ降解效果减弱的原因可能为nZVI颗粒活化PMS时表面发生氧化作用,Fe0含量降低,生成自由基速率减慢。Fe0反应生成的Fe2+和Fe3+在Fe0表面形成氧化层FexOy,该氧化层一方面阻止Fe0活化PMS反应,另一方面生成FexOy对PMS有一定催化活性[21]。试验说明,nZVI/PMS体系降解ATZ时,nZVI可重复使用,并保持较好催化活性。
4 结论
本文以实验室模拟ATZ污染水体为研究对象,以纳米零价铁活化过硫酸盐(nZVI/PMS)高级氧化工艺为手段,首先使用液相还原法成功合成nZVI并对其作SEM和XPS表征;进一步结合自由基淬灭试验分析nZVI/PMS体系降解ATZ反应机理;探究不同尺寸零价铁对ATZ降解效果的影响;最后探讨nZVI回收利用可能性。本研究得到如下结论:
a.液相还原法可成功制备纳米尺寸零价铁,该方法制备的nZVI颗粒呈球状,由于表面张力和磁性影响呈团聚现象。反应后nZVI颗粒表面被氧化呈不规则片状。
b.·SO4-、·OH均为nZVI/PMS体系中氧化活性物种;nZVI对ATZ无明显吸附降解作用。
c.nZVI/PMS体系可降解ATZ并取得良好效果,ATZ降解效果受零价铁尺寸影响,纳米零价铁活化PMS能力强于微米零价铁。
d.反应结束后nZVI固体颗粒可回收再次用于活化PMS降解ATZ试验;第3次使用仍具有良好催化效能。
