不同干燥方式对淫羊藿黄酮类成分含量及抗氧化活性影响
2021-07-21王燕萍贾旭森王子夏徐巧红史文博胡芳弟
王燕萍,贾旭森,王 艳,周 静,王子夏,李 雪,张 菊,徐巧红,史文博,胡芳弟
兰州大学药学院,甘肃 兰州 730000
淫羊藿为小檗科植物淫羊藿Epimedium brevicomuMaxim.、箭叶淫羊藿E.sagittatum(Sieb.et Zucc.) Maxim.、柔毛淫羊藿E.pubescensMaxim.或朝鲜淫羊藿E.koreanumNakai的干燥叶[1]。现代药理学研究表明,淫羊藿具有增强免疫[2]、抑制肿瘤[3-4]、抗炎[5]、抗骨质疏松[6-8]和抗氧化[9]等多种药理活性。目前,淫羊藿在临床上常用于治疗肾阳虚衰、阳痿遗精等症,上述功效与氧化应激高度相关。肾损伤患者因血清中氧化物活性增加且抗氧化物活性降低而导致氧化应激反应增加,进而导致肾功能障碍的发生[10-11],有研究表明淫羊藿总黄酮可以通过改善氧化应激,减少炎症反应,并通过抗凋亡途径来减少顺铂引起的小鼠肾损伤[12]。
淫羊藿的黄酮成分朝藿定A、朝藿定B、淫羊藿苷等在清除自由基、防止生物体过氧化过程中具有重要作用[13-14],可作为天然抗氧化剂,且朝藿定A、朝藿定B、朝藿定C和淫羊藿苷为《中国药典》2020年版中淫羊藿药材的指标性成分,说明上述黄酮是淫羊藿的重要活性成分。淫羊藿黄酮类成分的含量因品种[15]、产地[15]、种植模式[16]和采收时间[17]等不同而具有较大的差异,而采收后的干燥过程也会对其黄酮类成分产生较大的影响[18]。淫羊藿历来多以野生品供药用,药农采集药材后,随手扎把悬挂于山坡树叉上或房前屋后自然阴干或晒干,存在耗时长、较难规模化生产等缺点。有研究采用不同温度对朝鲜淫羊藿鲜叶进行了干燥,发现干燥温度与异戊二烯基黄酮类成分的含量呈反比[18]。
本实验采用不同干燥方式对淫羊藿鲜叶进行干燥,以黄酮类成分和醇浸出物含量以及抗氧化活性为指标,优选淫羊藿的最佳干燥方式,为其进一步开发利用提供理论依据。
1 仪器与材料
CPA225D十万分之一分析天平,德国赛多利斯科学仪器(北京)有限公司;FA2004万分之一分析天平,上海良华仪器仪表有限公司;安捷伦1260Ⅱ高效液相色谱仪,配有DAD检测器,安捷伦科技(中国)有限公司;KQ-400KDE超声波清洗仪,常州诺基仪器有限公司;UV-1700紫外分光光度计,日本Shimadzu公司。
淫羊藿鲜药材,来源于甘肃省礼县中坝乡,为野生品,5月初采集,采集后置于有冰袋的泡沫箱中,24 h内送至实验室进行相关实验,由甘肃中医药大学附属医院杨锡仓主任药师鉴定为小檗科淫羊藿属植物淫羊藿E.brevicomuMaxim.的叶。
对照品朝藿定A、朝藿定B、朝藿定C、淫羊藿苷和宝藿苷I,批号分别为110623-72-8、110623-73-9、110642-44-9、489-32-7和113558-15-9,质量分数均为98%,购自宝鸡市辰光生物科技有限公司;甲醇、乙腈,色谱纯,瑞典Oceanpak公司;其他试剂均为分析纯。
2 方法及结果
2.1 样品制备
称取500 g淫羊藿鲜叶36份,分别用不同温度(50、75、100、125、150、175、200 ℃)烘干、冷冻干燥、自然阴干、微波干燥、炒干、蒸后阴干,制得样品S1~S12,每个样品平行3份。样品信息见表1。

表1 淫羊藿不同干燥方式及所需时间Table 1 Different drying methods and required time of Epimedii Folium
2.2 水分含量测定
参照《中国药典》2020年版四部项下0832烘干法测定[19],结果通过Excel 2013进行处理,数据以表示(下同),结果见表2。《中国药典》2020年版规定淫羊藿的水分不得高于12%,根据表2,不同干燥方式制得的淫羊藿样品水分均低于12%,满足药典要求。
表2 水分测定结果 (±s , n = 3)Table 2 Moisture measurement results (±s , n = 3)

表2 水分测定结果 (±s , n = 3)Table 2 Moisture measurement results (±s , n = 3)
编号 含水量/% 编号 含水量/% 编号 含水量/%S1 7.07±0.14 S5 5.31±0.14 S9 5.52±0.40 S2 6.83±0.37 S6 4.73±0.07 S10 4.43±0.03 S3 6.53±0.11 S7 3.68±0.07 S11 6.31±0.20 S4 5.82±0.18 S8 5.82±0.92 S12 7.47±0.01
2.3 5种黄酮类成分含量测定
2.3.1 供试品溶液的制备 参照《中国药典》2020年版“淫羊藿”项下总黄酮醇苷的制备方法[1]。
2.3.2 对照品溶液的制备 分别取5种黄酮对照品适量,加甲醇制成对照品储备液(朝藿定A、朝藿定B、朝藿定C、淫羊藿苷和宝藿苷I的质量浓度分别为2.95、2.70、2.50、2.48、2.58 mg/mL),取5种对照品储备液各2 mL,置于10 mL量瓶中,制成混合对照品溶液。
2.3.3 色谱条件 色谱柱为Agilent Zorbax Eclipse XDB-C18柱(250 mm×4.6 mm,5 μm);柱温30 ℃;进样量10 μL;检测波长270 nm;流动相为水-乙腈,洗脱程序参照《中国药典》2020年版淫羊藿项下总黄酮醇苷的测定方法[1],具体为0~29 min,25%乙腈;29~30 min,25%~41%乙腈;30~55 min,41%乙腈。色谱图见图1。
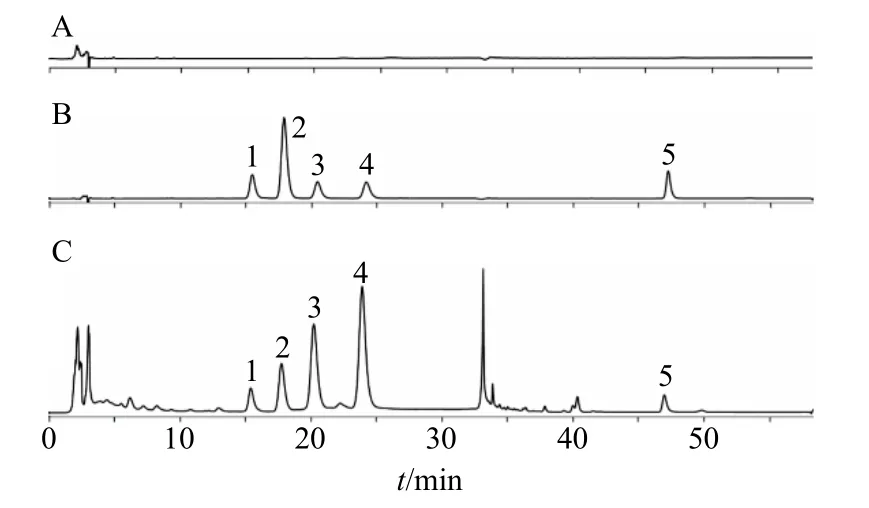
图1 空白溶剂 (A)、混合对照品 (B) 和淫羊藿样品 (C)的HPLC图Fig.1 HPLC of blank solvent (A), mixed reference substances (B), and Epimedii Folium sample (C)
2.3.4 线性关系考察 将上述混合对照品溶液稀释成系列对照品溶液(朝藿定A:1.15、2.30、4.61、9.22、18.44、36.88、73.75、147.50、295.00、590.00 μg/mL;朝藿定B:1.05、2.11、4.22、8.44、16.88、33.75、67.50、135.00、270.00、540.00 μg/mL;朝藿定C:0.98、1.95、3.91、7.81、15.63、31.25、62.50、125.00、250.00、500.00 μg/mL;淫羊藿苷:0.97、1.94、3.88、7.75、15.50、31.00、62.00、124.00、248.00、496.00 μg/mL;宝藿苷I:1.01、2.01、4.02、8.05、16.09、32.19、64.38、128.75、257.50、515.00 μg/mL),注入液相色谱仪10 μL,根据峰面积绘制标准曲线,进行线性回归,得5种成分的回归方程与线性范围分别为朝藿定AY=1 150.9X-137.38,R2=0.999 3,线性范围2.30~590.00 μg/mL;朝藿定BY=1 341.6X-171.33,R2=0.999 2,线性范围2.11~540.00 μg/mL;朝藿定CY=1 190.4X-122.67,R2=0.999 3,线性范围1.95~500.00 μg/mL;淫羊藿苷Y=1 593.3X-193.31,R2=0.999 0,线性范围1.94~496.00 μg/mL;宝藿苷IY=2 258.3X-148.44,R2=0.999 5,线性范围1.01~515.00 μg/mL。
2.3.5 样品测定 取各供试品溶液10 μL注入高效液相色谱仪,根据回归方程计算各黄酮类成分含量,结果见表3。《中国药典》2020年版规定淫羊藿药材中4种黄酮类成分朝藿定A、朝藿定B、朝藿定C和淫羊藿苷的含量和不得少于1.5%,本实验中除高温烘干样品(S5~S7)中4种黄酮含量和不满足药典要求外,其他样品中4种黄酮含量均符合要求。相比于阴干样品,微波干燥样品(S10)的4种黄酮总量最高,达到了4.00%。
表3 5种黄酮类成分测定结果 (±s , n = 3)Table 3 Determination results of five kinds of flavonoids (±s , n = 3)

表3 5种黄酮类成分测定结果 (±s , n = 3)Table 3 Determination results of five kinds of flavonoids (±s , n = 3)
*表示《中国药典》2020年版中淫羊藿药材的指标性成分朝藿定A~C和淫羊藿苷4种黄酮的总量*denote the total amount of for flavonoids, epimedin A—C and icariin, which are the indicator components of Epimedii Folium in the Chinese Pharmacopoeia (2020)
编号 朝藿定A/% 朝藿定B/% 朝藿定C/% 淫羊藿苷/% 4种黄酮总量*/% 宝藿苷I/% 5种黄酮总量/%S1 0.24±0.01 0.27±0.00 0.76±0.01 0.52±0.01 1.79±0.02 0.72±0.01 2.51±0.02 S2 0.31±0.01 0.35±0.01 1.26±0.03 0.85±0.01 2.78±0.04 0.62±0.01 3.39±0.03 S3 0.29±0.00 0.33±0.00 1.11±0.02 0.76±0.00 2.49±0.02 0.59±0.00 3.09±0.02 S4 0.29±0.01 0.34±0.00 1.04±0.04 0.76±0.05 2.42±0.08 0.61±0.01 3.04±0.29 S5 0.00±0.00 0.16±0.00 0.00±0.00 0.28±0.02 0.44±0.08 1.83±0.05 2.27±0.07 S6 0.00±0.00 0.15±0.00 0.00±0.00 0.28±0.00 0.43±0.01 2.47±0.17 2.90±0.16 S7 0.00±0.00 0.15±0.00 0.00±0.00 0.23±0.01 0.39±0.01 2.18±0.03 2.56±0.04 S8 0.26±0.00 0.31±0.00 0.97±0.02 0.69±0.02 2.23±0.02 0.78±0.01 3.01±0.03 S9 0.32±0.01 0.37±0.01 1.45±0.05 0.99±0.00 3.13±0.05 0.50±0.00 3.63±0.03 S10 0.35±0.01 0.40±0.01 1.90±0.03 1.34±0.08 4.00±0.08 0.44±0.01 4.43±0.18 S11 0.26±0.01 0.30±0.00 0.90±0.02 0.69±0.00 2.15±0.02 0.76±0.02 2.91±0.12 S12 0.28±0.00 0.33±0.00 0.97±0.01 1.35±0.02 2.91±0.03 1.06±0.02 3.98±0.05
考虑到宝藿苷I也是淫羊藿中重要黄酮类成分,也具有抗氧化[24]、抗骨质疏松[25]等活性,因此本实验将宝藿苷I的含量也作为评价指标之一。高温烘干样品(S5~S7)和蒸制后阴干样品(S12)中宝藿苷I含量较高,均达到了1.0%以上。
综合考虑4种黄酮类成分总量和宝藿苷I(5种黄酮)的含量,发现微波干燥样品(S10)中5种黄酮总量最高,达到了4.43%。不同干燥方式所得淫羊藿样品中5种黄酮类成分含量差异较大,可能与黄酮苷类成分受水解酶的影响在干燥过程中发生水解反应有关。
2.4 总黄酮含量测定[1]
2.4.1 供试品溶液制备 具体方法同“2.3.1”项。
2.4.2 对照品溶液的制备 取淫羊藿苷对照品2.50 mg,加甲醇制成0.500 mg/mL的对照品储备液,分别取对照品储备液0、100、200、300、400、500 μL,置于10 mL量瓶中,加甲醇定容至刻度,得系列质量浓度的淫羊藿苷对照品溶液。
2.4.3 线性关系考察 以空白溶剂校零,在270 nm波长下测定淫羊藿苷系列对照品溶液的吸光度(A)值,测定3次,取平均值,以淫羊藿苷对照品的质量浓度为横坐标(X),A值为纵坐标(Y),绘制标准曲线,进行线性回归,得回归方程Y=36.877X-0.027 5,R2=0.996 0,线性范围为0.005~0.025 mg/mL。
2.4.4 样品测定 精密吸取供试品溶液50 μL,置于5 mL量瓶中,加甲醇定容至刻度,混匀。测定A值,平行3份,根据A值计算总黄酮含量,结果见表4。《中国药典》2020年版规定淫羊藿的总黄酮含量不得少于5.0%,根据表4,不同干燥方式制得的12个淫羊藿样品中总黄酮含量均满足药典要求,总黄酮在(9.06±0.15)%~(10.39±0.11)%,其中冷冻干燥样品(S8)和微波干燥样品(S10)的总黄酮含量较高。
2.5 醇浸出物含量测定
按《中国药典》2020年版四部2201项下的热浸法测定[19],结果见表4。不同干燥方式所得样品中醇浸出物质量分数在(16.25±0.57)%~(29.72±0.13)%,高温烘干样品(S5~S7)的醇浸出物含量均低于20%,冷冻干燥样品(S8)的醇浸出物含量最高,达到29.72%。
表4 总黄酮和醇浸出物含量 (±s , n = 3)Table 4 Total flavones and alcohol extract contents (±s ,n = 3)
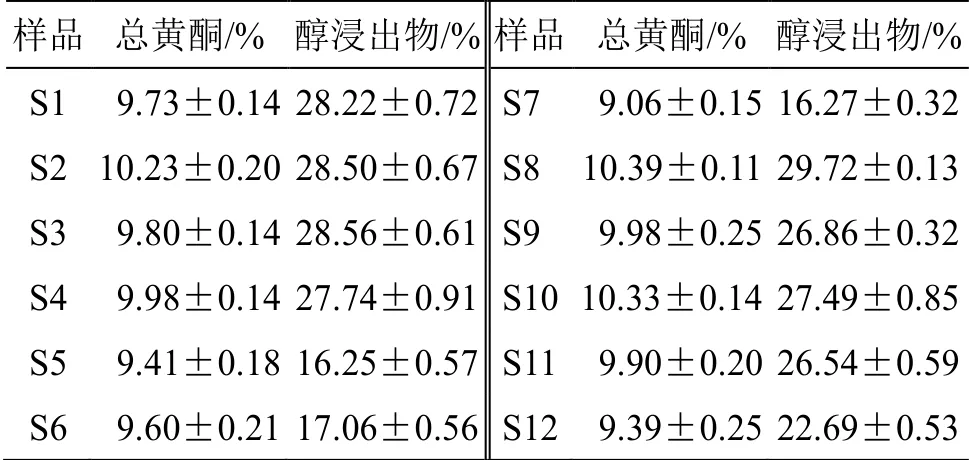
表4 总黄酮和醇浸出物含量 (±s , n = 3)Table 4 Total flavones and alcohol extract contents (±s ,n = 3)
样品 总黄酮/% 醇浸出物/% 样品 总黄酮/% 醇浸出物/%S1 9.73±0.14 28.22±0.72 S7 9.06±0.15 16.27±0.32 S2 10.23±0.20 28.50±0.67 S8 10.39±0.11 29.72±0.13 S3 9.80±0.14 28.56±0.61 S9 9.98±0.25 26.86±0.32 S4 9.98±0.14 27.74±0.91 S10 10.33±0.14 27.49±0.85 S5 9.41±0.18 16.25±0.57 S11 9.90±0.20 26.54±0.59 S6 9.60±0.21 17.06±0.56 S12 9.39±0.25 22.69±0.53
2.6 指纹图谱研究
2.6.1 供试品溶液制备 同“2.3.1”项。
2.6.2 色谱条件 色谱柱为Agilent Zorbax Eclipse XDB-C18(250 mm×4.6 mm,5 μm);柱温20 ℃;体积流量1 mL/min;进样量20 μL;检测波长270 nm;流动相为0.5%乙酸水溶液-甲醇-乙腈;洗脱程序参考文献报道[20],稍作调整,具体见表5。
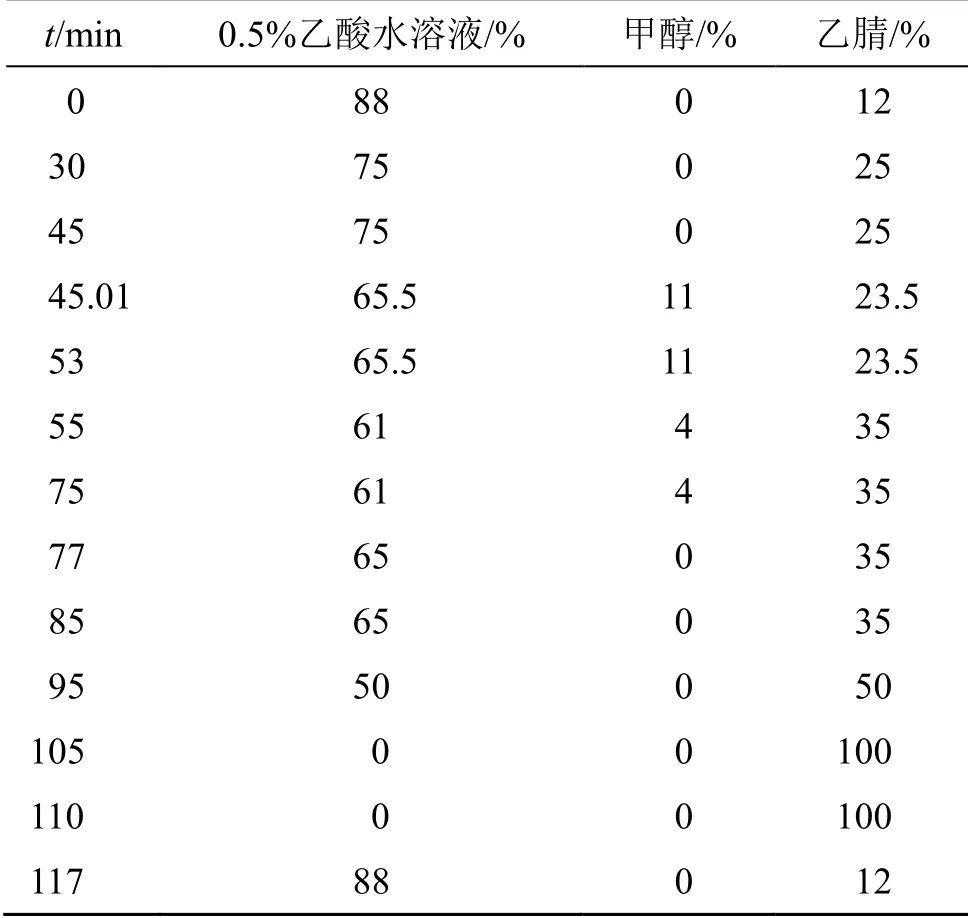
表5 梯度洗脱程序Table 5 Gradient elution program
2.6.3 指纹图谱的建立及结果分析 按“2.6.2”项下色谱条件测定,得12批不同干燥方式所得淫羊藿样品的指纹图谱。本实验共标定了12个共有峰,通过与对照品比对(图2),指认出了其中的3个,分别为朝藿定B(5号峰),淫羊藿苷(6号峰)和宝藿苷I(12号峰)。将cdf格式的数据导入《中药色谱指纹图谱相似度评价系统》2004A版进行分析,设置S9(阴干样品)为参照指纹图谱,采用多点校正后进行匹配,12批样品的指纹图谱叠加图见图3。通过图3可以看出不同干燥方式对化学成分的影响较大,高温烘干样品(S5~S7)在0~50 min共有峰较少且峰面积较小,50 min以后共有峰峰面积增加,其中峰12(宝藿苷I)增幅最大,在50~55 min,出现了1个新峰X。猜测淫羊藿在高温处理过程中存在着黄酮糖苷之间的转化,由多糖苷(朝藿定A、朝藿定B、朝藿定C和淫羊藿苷)脱去了C7位的葡萄糖,转化为了低糖苷(箭藿苷A、箭藿苷B、鼠李糖基淫羊藿次苷II和宝藿苷I),以淫羊藿苷转化为宝藿苷I为例,转化过程见图4,查阅相关文献可证实淫羊藿在炮制过程中黄酮糖苷之间确实存在转化的现象[26-27],转化机制有待进一步研究。

图2 空白溶剂 (A)、混合对照品 (B)、阴干样品 (C) 和200 ℃烘干样品 (D) 的HPLC图Fig.2 HPLC of blank solvent (A), mixed reference substances (B), shade drying sample (C) and sample dried at 200 ℃ (D)
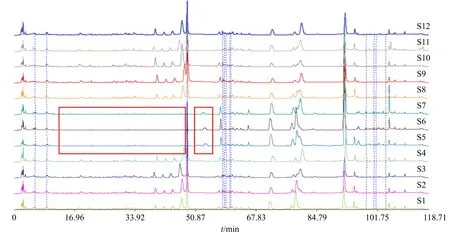
图3 不同干燥方式指纹图谱比较Fig.3 Comparison of fingerprint of different drying modes

图4 淫羊藿苷转化为宝藿苷IFig.4 Transformation of icariin into baohuoside I
将12个样品共有峰的比值和进行归一化处理,结果见表6。可见,高温烘干(S5~S9)样品的指纹图谱中共有峰的比值和较高,其中175 ℃烘干样品(S6)的共有峰的比值和最高。
表6 指纹图谱共有峰峰面积与称样量比值和 (±s , n = 3)Table 6 Sum of ratio of common peak area to sample weight in fingerprint (±s , n = 3)

表6 指纹图谱共有峰峰面积与称样量比值和 (±s , n = 3)Table 6 Sum of ratio of common peak area to sample weight in fingerprint (±s , n = 3)
样品 共有峰峰面积与称样量的比值和(归一化) 样品 共有峰峰面积与称样量的比值和(归一化)S1 0.96±0.02 S7 1.53±0.01 S2 1.04±0.02 S8 1.03±0.06 S3 0.97±0.02 S9 1.00±0.05 S4 0.98±0.04 S10 1.10±0.04 S5 1.37±0.03 S11 1.03±0.03 S6 1.65±0.05 S12 1.16±0.00
2.7 综合评分分析及结果
参照罗春丽等[23]采用综合评分法,评价不同干燥方式对黄酮类成分含量的综合影响。根据所测成分在淫羊藿中的重要性,以总黄酮醇苷(以5种黄酮总含量计)为40分,总黄酮含量为30分,醇浸出物含量为10分,比值和为20分,满分100分,每个样品的综合得分=(5种黄酮含量和/最大5种黄酮含量和)×40+(总黄酮含量/最大总黄酮含量)×30+(醇浸出物含量/最大醇浸出物含量)×10+(比值和/最大比值和)×20。综合得分结果见表7,可以看出,微波干燥样品(S10)的综合得分最高,达到了92.50,因此,通过黄酮类成分含量优选的干燥方式为微波干燥。

表7 综合得分Table 7 Composite scores
2.8 抗氧化活性测定
2.8.1 供试品溶液制备 同“2.3.1”项。
2.8.2 测定方法
(1)DPPH自由基清除实验[21-22]:精密移取1 mL稀释10倍后的供试品溶液与1 mL 80 μg/mL DPPH溶液于试管中,摇匀,避光反应30 min,以蒸馏水为空白调零,在517 nm波长下测定A值(A1)。用稀乙醇代替供试品,测定A值(A0),根据公式自由基清除率=(1-A1/A0)计算DPPH自由基清除率,每个样品重复3份,求平均值。
(2)羟自由基清除实验[21-22]:移取供试品溶液0.2 mL,依次加入反应液[20 mmol/L pH 7.4的磷酸盐缓冲液,2.67 mmol/L脱氧核糖,100 mmol/L乙二胺四乙酸(EDTA)]0.6 mL、0.4 mmol/L的硫酸亚铁胺溶液0.2 mL、10 mmol/L过氧化氢溶液0.2 mL,在37 ℃水浴加热15 min,再分别加入1%的硫代巴比妥酸(TBA)溶液和2%的三氯乙酸(TCA)溶液各1 mL,终止反应,然后沸水浴加热15 min后冷却至室温,以蒸馏水为空白调零,在532 nm下测定A值(A1),用稀乙醇代替供试品,测定A值(A0),根据公式计算,每个样品重复3份,求平均值。
(3)超氧阴离子自由基清除实验[21-22]:超氧自由基在吩嗪硫酸甲酯(PMS)-还原型辅酶I(NADH)体系中生成,用于氯化氮蓝四唑(NBT)还原的检测。取供试品溶液1 mL,加入Tris-HCl缓冲液(16 mmol/L,pH 8.0)、NADH-2Na(338 mmol/L)、PMS(30 mmol/L)、NBT(72 mmol/L)各1 mL,混匀后,在室温反应5 min,用蒸馏水校零,在560 nm处测定A值(A1),用稀乙醇代替供试品,测定A值(A0),根据计算,每个样品重复3份,求平均值。
2.8.3 抗氧化活性结果及谱效关联分析 不同干燥方式所得样品的稀乙醇提取液的自由基清除能力有较大差异,其中微波干燥样品(S9)的稀乙醇提取物对DPPH自由基和超氧阴离子自由基的清除能力较强,高温烘干样品(S5~S7)的稀乙醇提取物对羟自由基的清除能力较强,结果见表8。为了进一步阐述上述结果,本实验采用偏最小二乘法(partial least square method,PLS)对12个样本的指纹图谱和自由基清除活性进行了相关性分析,以各样品对3种自由基的清除率为因变量(Y),以各批样品醇提物指纹图谱中共有峰的峰面积与称样量的比值为自变量(X)进行拟合,结果见图5。12个共有峰中有6个共有峰与DPPH自由基清除率呈正相关,分别为峰1、2、5~7、12,其中峰5、6、12分别为朝藿定B、淫羊藿苷和宝藿苷I。5个共有峰与羟自由基清除率呈正相关,分别为峰7~11。7个共有峰与超氧阴离子自由基清除率呈正相关,分别为峰1~3、5、6、12,其中峰5、6、12分别为朝藿定B、淫羊藿苷和宝藿苷I。谱效关联结果表明,与DPPH和超氧阴离子自由基清除活性呈正相关的成分主要集中在极性较大的部分,其中朝藿定B、淫羊藿苷均是与DPPH和超氧阴离子自由基清除活性呈正相关的成分,而微波干燥样品中这2个成分较高,进一步证明了优选工艺的合理性;而与羟自由基清除活性成正相关的主要色谱峰多分布在极性较小的部分,高温烘干样品(S5~S7)的极性较小部分含量较高,因此,高温烘干样品的羟自由基清除能力相对较高。

图5 谱效关联分析DPPH自由基 (A)、羟自由基 (B) 和超氧阴离子自由基 (C) (±s , n = 3)Fig.5 Spectral correlation analysis of DPPH radical (A),hydroxyl radical (B) and superoxide anion radical (C)(±s , n = 3)
表8 淫羊藿稀乙醇提取液清除自由基活性比较 (±s,n = 3)Table 8 Comparison of free radical scavenging activities of Epimedii Folium dilute ethanol extract (±s , n = 3)
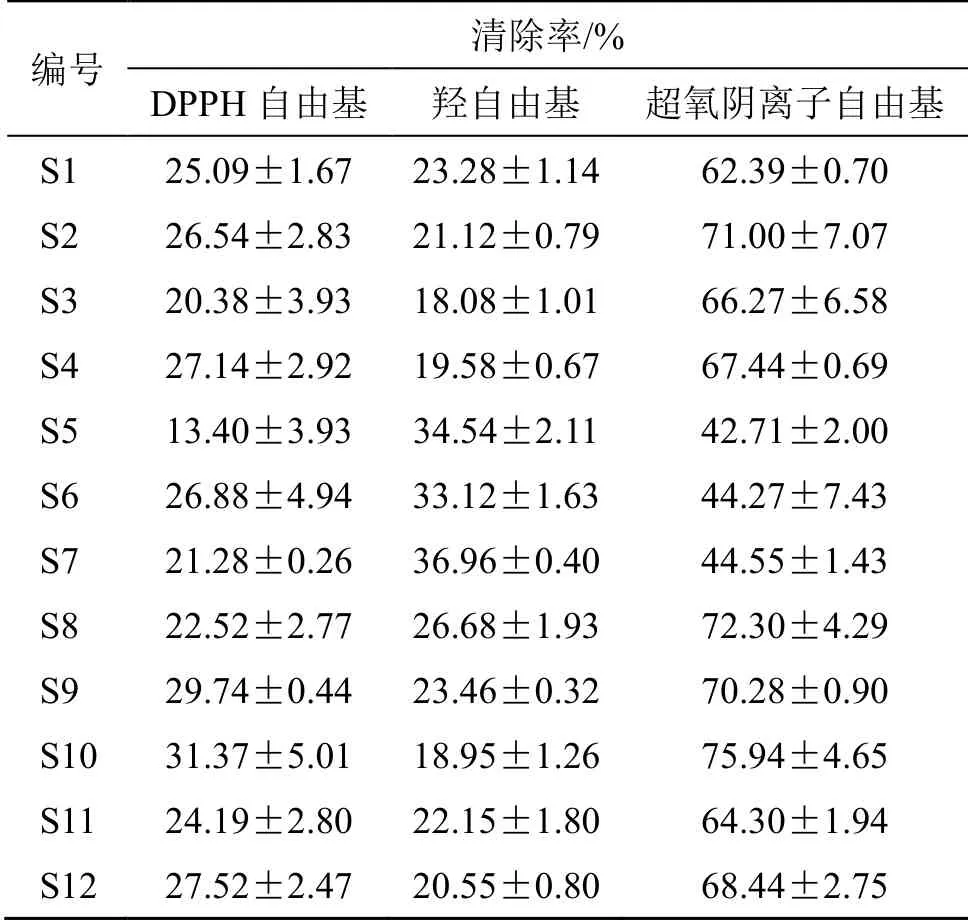
表8 淫羊藿稀乙醇提取液清除自由基活性比较 (±s,n = 3)Table 8 Comparison of free radical scavenging activities of Epimedii Folium dilute ethanol extract (±s , n = 3)
编号 清除率/%DPPH自由基 羟自由基 超氧阴离子自由基S1 25.09±1.67 23.28±1.14 62.39±0.70 S2 26.54±2.83 21.12±0.79 71.00±7.07 S3 20.38±3.93 18.08±1.01 66.27±6.58 S4 27.14±2.92 19.58±0.67 67.44±0.69 S5 13.40±3.93 34.54±2.11 42.71±2.00 S6 26.88±4.94 33.12±1.63 44.27±7.43 S7 21.28±0.26 36.96±0.40 44.55±1.43 S8 22.52±2.77 26.68±1.93 72.30±4.29 S9 29.74±0.44 23.46±0.32 70.28±0.90 S10 31.37±5.01 18.95±1.26 75.94±4.65 S11 24.19±2.80 22.15±1.80 64.30±1.94 S12 27.52±2.47 20.55±0.80 68.44±2.75
3 讨论
本实验将淫羊藿鲜药材采用不同干燥方式处理,制得12个样品,通过测定指标性成分含量,采用综合评分法初步筛选最佳的干燥方式,再通过比较不同样品对3种自由基的清除能力,综合黄酮类成分含量和抗氧化活性优选最佳的干燥方式。
本实验选择评价指标时,首先选取了药典中淫羊藿项下的4种指标性成分朝藿定A、B、C和淫羊藿苷,考虑到宝藿苷I是淫羊藿苷在体内的主要代谢产物[28-29],在抗氧化[24]、抗骨质疏松[25]等方面具有显著的生物活性。相比与二糖苷淫羊藿苷,单糖苷宝藿苷I具有更强的抗氧化活性[24],且极性较小,在体内更容易通过细胞膜。因此,将宝藿苷I的含量也作为指标性成分进行考察,因此,本实验重点关注了上述5种黄酮的含量。淫羊藿的主要成分为黄酮类,总黄酮也是药典规定的评价指标之一,而其醇浸出物在一定程度上也能反映黄酮的含量,因此,将总黄酮含量和醇浸出物含量也作为评价指标。指纹图谱中的共有峰能够从整体、定量地把握样品的质量,因此将比值和也作为评价指标之一。
以上述4个指标通过综合评分法评价,发现微波干燥样品的综合得分最高,因此,通过黄酮类成分的含量优选的干燥方式为微波干燥。体外自由基清除实验证明微波干燥样品对DPPH自由基和超氧阴离子自由基的清除能力均最强;高温烘干样品对羟自由基的清除能力较高。谱效关联结果表明与DPPH和超氧阴离子自由基清除活性呈正相关的成分主要集中在极性较大部分,其中朝藿定B、淫羊藿苷均是与DPPH和超氧阴离子自由基清除活性呈正相关的成分,而微波干燥样品中这2个成分较高,进一步证明了优选工艺的合理性。
综上所述,微波干燥样品中黄酮类成分、醇浸出物含量较高,且微波干燥样品的稀乙醇提取物对DPPH自由基和超氧阴离子自由基的清除活性较强,综合考虑黄酮类成分含量及抗氧化活性,建议最佳的干燥方式为微波干燥,但是微波干燥需要特殊的设备,因此,此方法是否能用于淫羊藿工业化生产的干燥步骤,还有待进一步研究。
利益冲突所有作者均声明不存在利益冲突
