基于氧化还原响应型刺芒柄花素印迹的智能药物载体研究
2021-07-21刘雅晴
谭 倪,陈 灿,廖 森,刘雅晴,丁 醉
(南华大学 化学化工学院,湖南 衡阳 421001)
0 引 言
刺芒柄花素(formononetin)属于异黄酮类化合物,其主要来源于豆科植物红车轴草的花序及带花枝叶、刺芒花全草,是当归、黄芪、葛根、鸡血藤等常用中草药的活性成分之一[1]。刺芒柄花素结构式如图1所示,由于具有类似于动物体内雌激素样的双羟基酚式结构,所以较高剂量的刺芒柄花素在体内便具有雌激素样作用,它可在一定程度上改善妇女更年期症状,对生殖系统具有一定的安全性[2]。近来,国内外研究还表明,刺芒柄花素具有抗氧化、抗动脉粥样硬化、预防骨质疏松、乳腺癌等效用[3-6]。
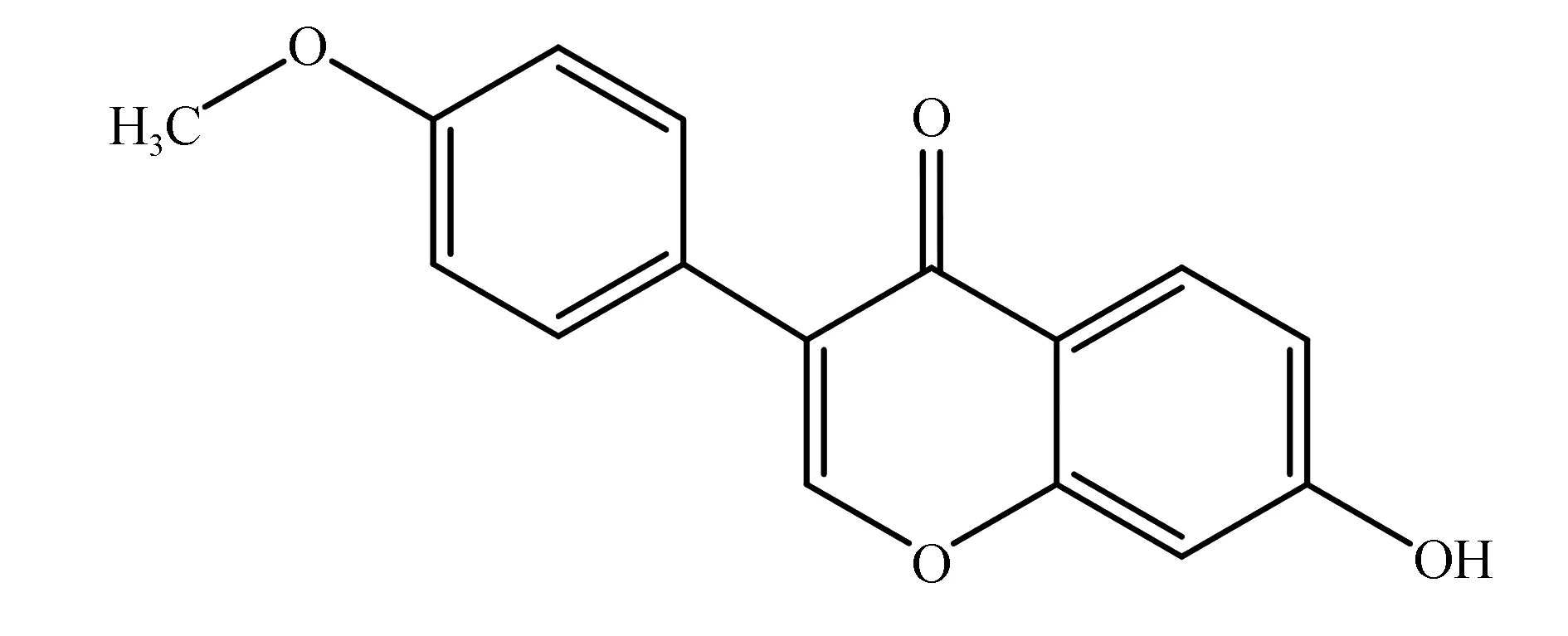
图1 刺芒柄花素结构式
癌症作为一种“健康杀手”,它严重威胁着人们的生命,因此,对于癌症的预防治疗一直便是国内外学者研究的热点。目前,化学治疗法是临床上治疗恶性肿瘤最常用的方法,该法虽能杀灭患者体内的癌细胞,实现全身性治疗,但它对机体正常组织细胞同样具有一定的损伤[7],产生多种副作用,临床主要表现为免疫力下降、头发脱落、恶心呕吐等。另外,当前许多用于化学治疗的抗肿瘤药物还具有溶解度低、生物利用率低、靶向性差等诸多不足。近年来,纳米载药系统不仅能够有效解决抗肿瘤药物水溶性差及药物在其他部位富集等问题、而且还能延缓药物体内释放速度,并对肿瘤微环境具有刺激性响应的载体可实现靶向释放。当前已经上市或进入临床阶段的肿瘤纳米制剂的载体有脂质体、聚合物胶束、白蛋白纳米粒等[8-10]。由于肿瘤细胞往往具有特殊的微环境[11-12],研究者还进一步开发设计了刺激响应型纳米药物载体,如pH响应型[13]、光响应型[14]、温度响应型[15]、多重响应型[16]等。由此可见,未来对环境响应性纳米药物载体的研究应具有十分广阔的前景。
本论文利用肿瘤细胞高还原状态环境,以刺芒柄花素为模板,甲基丙烯酸为功能单体,含二硫键的N,N′-双丙烯酰胱胺(BAC)为交联剂,Fe3O4@葡聚糖磁性纳米粒子(Fe3O4@Dextran)为基体制备了具有氧化还原响应的刺芒柄花素分子印迹聚合物,该聚合物对还原环境刺激敏感,生物毒性小且可降解,有望实现药物靶向释放,对抗氧化、抗乳腺癌等的智能给药系统研究具有一定意义。
1 材料与方法
1.1 试剂与仪器
试剂:甲基丙烯酸(MAA,98%)、刺芒柄花素(formononetin,98%)、五甲基二乙烯三胺(PMDETA,98%)、乙二醇二甲基丙烯酸酯(EGDMA,99%)、葡聚糖(Dextran,分子量2万)、大豆苷元(daidzein,98%)、N,N′-双丙烯酰胱胺(BAC、98%)、还原性谷胱甘肽(GSH,98%)、硫酸亚铁·七水(FeSO4·7H2O)、无水三氯化铁(FeCl3)和2,2′-偶氮异丁腈(AIBN),均为分析纯。
仪器:UV-8500分光光度计(天津市普瑞斯仪器有限公司)、7307型振动样品磁强计(美国Lakeshore有限公司)、MutliFlex 2kW X射线衍射仪(兴和仪器上海有限公司),Nicolet IS 10傅立叶红外光谱仪(美国Thermo fisher有限公司)。
1.2 氧化还原响应刺芒柄花素分子印迹聚合物的制备
1.2.1 磁性Fe3O4@Dextran载体的制备
根据先前的文献报道[17-18],本试验采用共沉淀法制备Fe3O4@Dextran:将0.002 mol FeSO4·7H2O和0.004 mol FeCl3溶于蒸馏水中,得澄明溶液备用。称取100 mg葡聚糖溶于50 mL体积分数为0.25%醋酸溶液,然后将之加入到上述溶液体系中,再缓慢加入10 mL NH4OH,在N2保护下,加热至75 ℃搅拌2 h,反应结束后冷却至室温,采用磁铁将产物Fe3O4@Dextran与反应溶液分离,甲醇洗涤至中性,-45 ℃下冷冻干燥。
1.2.2 氧化还原响应刺芒柄花素分子印迹聚合物的合成
基于课题组之前研究[19],采用反向原子转移自由基聚合法(reverse atom transfer radical polymerization,RATRP)合成目标产物。取0.026 8 g刺芒柄花素,超声溶于20 mL甲醇,再加入35.2 μL MAA,反应体系在N2氛围下,室温搅拌12 h。预聚合结束后,依次向反应体系中加入EGDMA(377 μL),BAC(0.130 0 g),AIBN(60 mg),CuBr2(10 mg),PMDETA(20.1 μL),Fe3O4@Dextran载体100 mg,在N2保护下,75 ℃恒温反应24 h。反应完成后,借助外磁场分离产物,甲醇洗涤除去表面附着物,随后将产物置于体积比为7:3的甲醇-乙酸洗脱液中,采用索氏提取法洗脱模板分子至检测不到紫外吸收,然后用乙醇反复洗涤,最后产物置于60 ℃真空干燥。非印迹聚合物(non-imprinted polymer,NIPs)合成不加入模板分子,其他步骤与MIP的合成一致。MIP合成路线如图2所示。

图2 刺芒柄花素分子印迹材料的制备过程图
1.3 印迹材料的吸附性能研究
1.3.1 静态吸附实验
称取3 mg MIP/NIP分别加入到5 mL一系列不同浓度刺芒柄花素溶液中,室温下震荡420 min,随后利用外磁场分离吸附剂,在260 nm处测定上清液吸光度,得到刺芒柄花素的吸附平衡浓度,根据公式(1)计算出两种材料的吸附量Qe。
(1)
式中:C0(mg/mL)和Ce(mg/mL)分别是刺芒柄花素初始质量浓度和平衡质量浓度;V(mL)代表吸附体系溶液的体积;M(g)为MIP或NIP的质量。
1.3.2 动力学吸附实验
分别称取MIP/NIP 3 mg于5 mL 0.200 mg/mL刺芒柄花素甲醇溶液中,室温下,混合物在振荡器中孵育10、20、40、80、120、180、240、300、420、540和660 min,磁铁分离吸附剂,取上清液过滤后,测量刺芒柄花素的吸光度,由方程(1)得到对应时间内的吸附量。
1.3.3 选择性考察实验
本实验采用结构类似的大豆苷元(daidzein)作为竞争底物探索材料的选择性。将3 mg MIP/NIP分别加入到0.200 mg/mL刺芒柄花素和大豆苷元溶液中,室温下震荡420 min,上清液过滤后,通过UV测定其紫外吸收,利用公式(2)~公式(4)得到MIP/NIP分别对刺芒柄花素和大豆苷元的吸附量及相应的选择性参数。
Kd=Qe/Ce
(2)
α=Kd1/Kd2
(3)
β=α1/α2
(4)
式中:Kd1和Kd2分别为刺芒柄花素和大豆苷元的分布系数;Ce(mg/mL)为刺芒柄花素和大豆苷元的平衡浓度;α指选择系数;β为MIP的相对选择系数。
1.3.4 重复再生性能研究
取3 mg MIP与5 mL 0.200 mg/mL的刺芒柄花素溶液混合,室温下震荡420 min后,外磁场分离上清液测定其吸光度,转移吸附剂至索氏提取器中,洗涤除去模板分子至检测不到紫外吸收,然后将吸附剂干燥后用于下一次吸附。重复上述吸附-解吸实验6次,计算每一循环的吸附量。
1.4 印迹材料还原刺激响应性释放实验
称取10 mg载药MIP/NIP分散于20 mL含有吐温80磷酸缓冲溶液(PBS,pH 7.4)、PBS(pH 7.4,10 mol/L GSH)中,混合体系在温度为37 ℃,转速150 r/min条件下体外释放。在预设时间点,取出3 mL上层释放介质,并加入同体积的新鲜缓冲溶液保持溶液体积不变,检测上清液的吸光度,然后根据公式(5)计算不同条件下药物累积释放量。
(5)
式中:Cn(mg/mL)表示第n次取样时刺芒柄花素溶液的质量浓度;V1(mL)为缓冲溶液的总体积;V(mL)代表置换溶液的体积;M(mg)指吸附剂的载药量。
2 结果与分析
2.1 表征分析
2.1.1 FT-IR分析

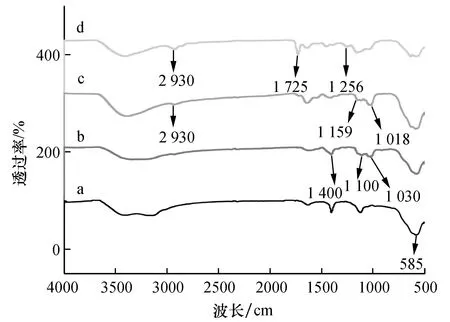
a—Fe3O4;b—Fe3O4@Dextran;c—MIP;d—吸附后MIP。
2.1.2 VSM分析
Fe3O4(曲线a),Fe3O4@Dextran(曲线b),MIP(曲线c)三种物质的磁回滞线如图4所示。室温条件下,Fe3O4,Fe3O4@Dextran,MIP的饱和磁化强度分别为81.75、70.93、45.25 Am2/kg。Fe3O4@Dextran,MIP磁饱和强度相比于Fe3O4依次降低的原因可能是葡聚糖包裹层和印迹层的顺次加入。同时,在原点处三种物质的矫顽力趋近于零,即没有出现磁滞现象。以上结果表明,三种粒子具有超顺磁性[23],制备的印迹材料表现出良好的磁响应性。

a—Fe3O4;b—Fe3O4@Dextran;c—MIP。
2.2 印迹材料的吸附性能研究
2.2.1 静态吸附研究
由吸附等温线图5可知,初始浓度小于0.200 mg/mL时,MIP对刺芒柄花素的吸附量与初始浓度呈正相关,吸附量随浓度的增加而增大,当浓度达到0.200 mg/mL时,吸附达到平衡状态,平衡吸附量为31.170 mg/g。这是由于随着浓度增大,MIP上的印迹孔穴利用率随之提高,当浓度增大至0.200 mg/g时,MIP表面的印迹位点基本达到饱和状态,故实现吸附平衡。在整个吸附过程中,NIP的吸附量明显低于MIP,其最大吸附量仅为7.167 mg/g,可见,MIP表面存在大量刺芒柄花素的特异性结合位点,因此对其具有更高的亲和力。以上结果表明,MIP对刺芒柄花素具有特异性吸附,而NIP对其仅具有非特异性吸附(物理吸附)。

图5 MIP,NIP的静态吸附曲线
随后,基于以上数据运用Scatchard模型对其进一步分析,探究两种材料的吸附特性。Scatchard方程式如下:
(6)
式中:Qe(mg/g)为MIP或NIP在不同浓度溶液中的吸附量;Qmax(mg/g)是最大表观结合量;C(mg/mL)表示刺芒柄花素吸附平衡时的浓度;Kd为平衡解离常数。
MIP/NIP的Scatchard拟合曲线如图6(a),(b)所示,相关参数如表1所示。图6(a)中存在两条斜率不同的直线,表明MIP对刺芒柄花素的结合是不均匀的,其内部可能存在两种不同类型的吸附位点[24],即高亲和位点与低亲和位点,其线性方程分别为:Y1=-11.452 9X+452.944 39,Y2=-7.612 6X+81.394 76,根据两条直线的截距和斜率,可得到高亲和位点和低亲和位点的Kd和Qmax分别为0.087 31 mg/mL和0.131 36 mg/g,及39.548 45 mg/g和10.692 11 mg/g;图6(b)中只出现了一条直线,且线性关系较差,这说明NIP表面对模板分子仅存在一种吸附类型,即为低亲和位点,其线性拟合方程为:Y3=-18.062 36X+138.011 59,计算得出Kd和Qmax分别为0.055 36 mg/mL和7.641 00 mg/g,说明NIP对刺芒柄花素是简单的物理结合,且结合能力较低。综上可知,相对于NIP,MIP对刺芒柄花素具有更强的吸附性能。

图6 MIP和NIP的Scatchard拟合图

表1 Scatchard拟合参数
2.2.2 动力学吸附研究
根据动态吸附图7可知,前期MIP对刺芒柄花素的吸附量随时间的增加而增大,增长速率较快,主要由于其表面具有大量特定印迹孔穴,可快速结合溶液中的模板分子,当吸附时间达到420 min后,吸附量达到最大值(31.170 mg/g)且趋向平衡,表明随时间推移,材料表面的印迹位点逐渐被模板分子占据,最终达到饱和状态。相比于MIP,在任一时间点,NIP对模板分子的吸附量都明显低于MIP,而且随时间增长较缓慢,在180 min便达到吸附平衡,平衡时间比MIP提前了140 min,此时的最大吸附量为7.167 mg/g,以上结果归因于,MIP内部存在大量特异性吸附位点,而NIP对模板分子不具有特异性吸附,仅为简单的物理吸附过程。
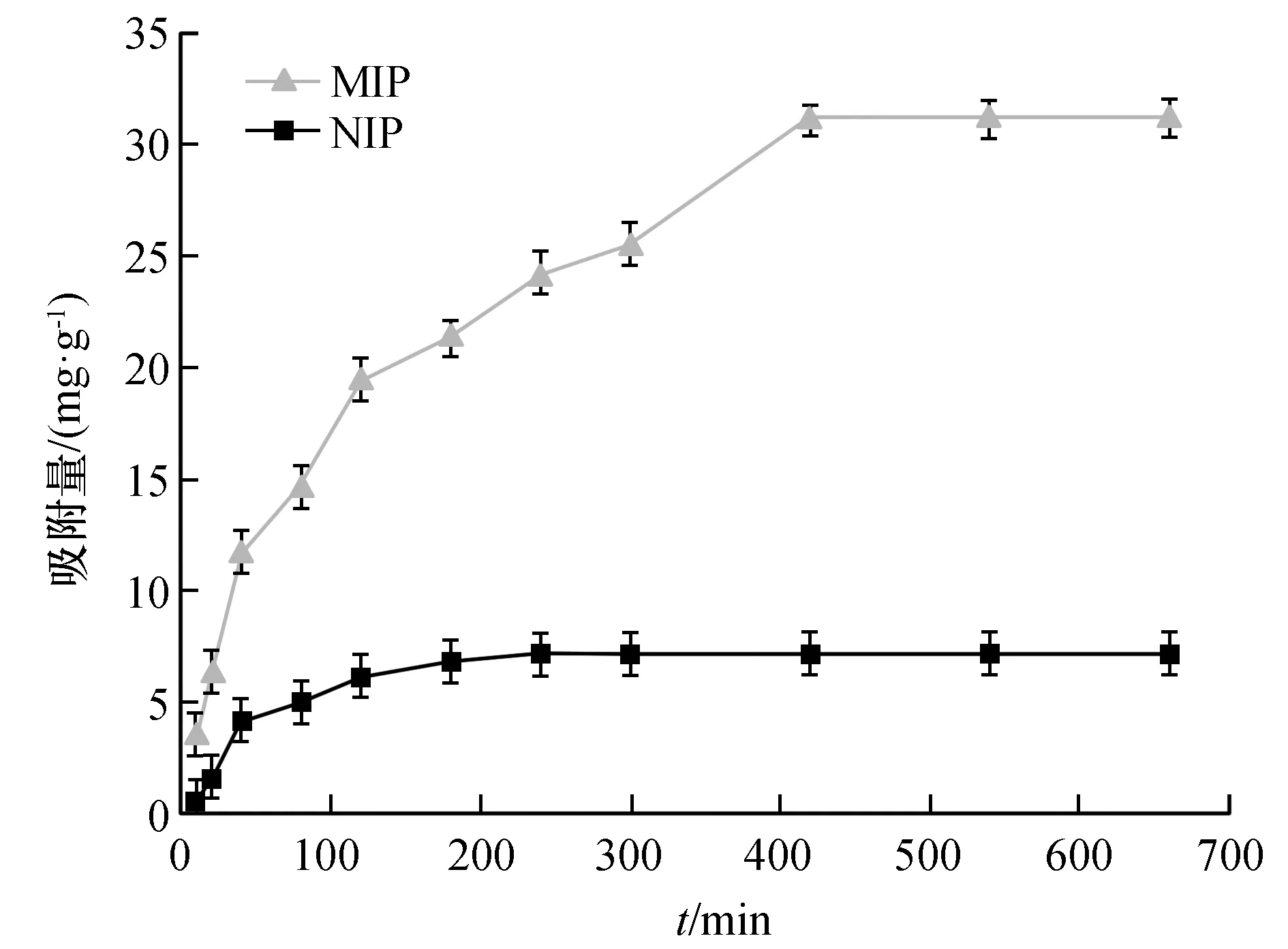
图7 MIP/NIP动力学吸附图
2.2.3 选择性研究
选择性实验结果如图8所示,MIP对刺芒柄花素的吸附量高达31.170 mg/g,而对大豆苷元的吸附量仅为刺芒柄花素的一半(15.000 mg/g);相比于MIP,NIP对模板分子和竞争底物的吸附量相差不大(7.167 mg/g,8.500 mg/g),由此可知,MIP对模板分子具有特异识别性。

图8 MIP/NIP对刺芒柄花素和大豆苷元的吸附量
随后,根据公式(2)~公式(4)得到两种材料的选择性相关参数,列于表2。由表可知,吸附剂为MIP时,对刺芒柄花素的分布系数Kd值(167.671 mg/mL)显然大于大豆苷元(58.820 mL/g),且MIP对两种物质的选择系数α为2.851,而NIP的选择系数仅为0.361;同时,MIP对两者的相对选择性因子β为7.898,远大于1。所以,MIP对刺芒柄花素具有高选择性和亲和力。

表2 选择性相关参数
2.2.4 重复再生性能研究
MIP再生性实验结果如图9所示。由图9可知,六次循环试验后,MIP对模板分子的吸附量由31.170 mg/g减少到27.167 mg/g,相比于初次吸附仅降低了12.84%,这可能是由于洗脱和干燥过程中,少量三维印迹空腔被破坏,特异性结合位点减少。可见,印迹材料保持较高的稳定性和较强重复利用性能。
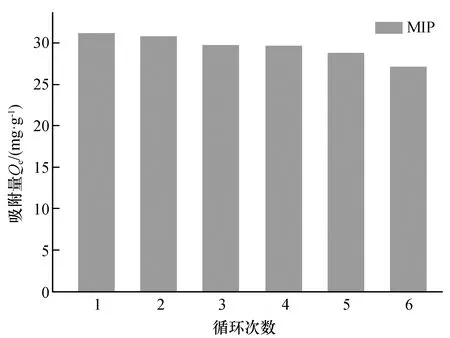
图9 MIP重复再生性能
2.3 印迹材料还原刺激响应性释放分析
MIP的还原响应性实验结果如图10所示,pH值为7.4的条件下,刺芒柄花素在含有GSH的溶液中累积释放量和释放时间大于不含GSH的对照组,其最大累积释放量达42.43%,释放时间为90 h;而在不含GSH的介质中最大释放量仅为10.09%,80 h达到释放平衡,二者累积释放量相差4倍;造成上述现象的主要原因是,在还原环境中,交联剂BAC中的S-S容易断裂,使材料的交联结构受到破坏,印迹孔穴坍塌,增大了药物的释放量,因此,MIP具有较强的还原敏感性,预计对肿瘤细胞微环境具有氧化还原响应。
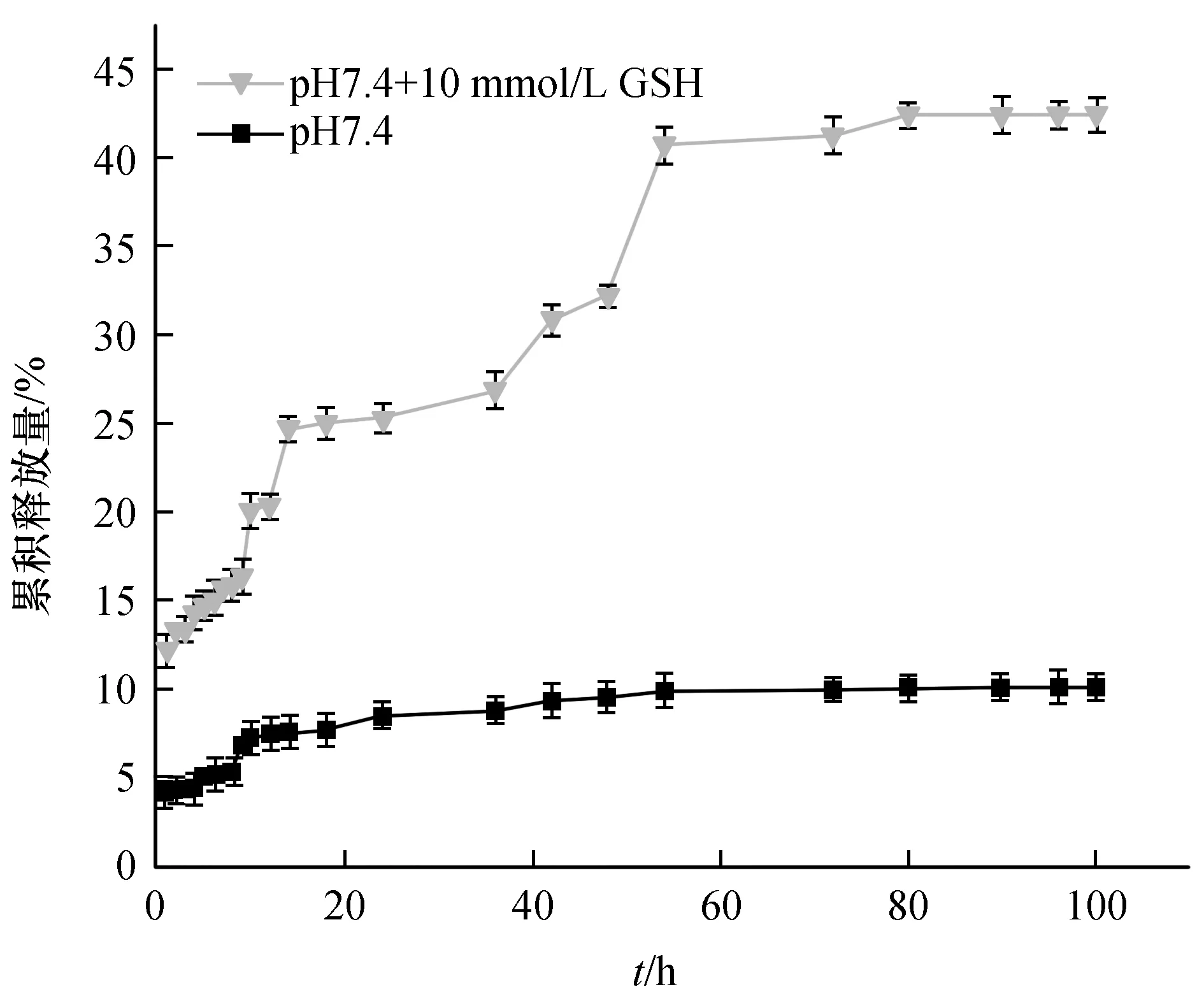
图10 载药MIP在不同环境中刺芒柄花素累积释放率
3 结 论
本论文以刺芒柄花素为模板分子,甲基丙烯酸为功能单体,引入含二硫键的N,N′-双丙烯酰胱胺为交联剂,运用反向原子自由基聚合法成功合成了具有氧化还原响应的刺芒柄花素分子印迹聚合物,该材料不仅对模板分子具有良好的吸附性能(优化条件下最大载药量达31.170 mg/g,是非印迹材料载药量的4.35倍),而且具有较强的选择性,其选择性因子(α)和相对选择性因子(β)分别为2.851,7.898。Scatchard分析结果表明印迹材料内部存在两种结合位点,分别是高亲和位点和低亲和位点。药物释放结果表明目标产物对氧化还原刺激具有较好响应性:还原条件下药物累积释放量是非还原条件下4倍。
