涝害对不同大豆品种根际微生物群落结构特征的影响
2021-07-19禹桃兵石琪晗连腾祥
禹桃兵 石琪晗 年 海 连腾祥
涝害对不同大豆品种根际微生物群落结构特征的影响
禹桃兵 石琪晗 年 海*连腾祥*
华南农业大学农学院/ 国家大豆改良中心广东分中心, 广东广州 510642
淹水影响不同大豆品种根际微生物群落组成, 不同基因型大豆植株耐涝性差异较大。本研究选取耐涝(waterlogging-tolerant, W-T)基因型大豆齐黄34和涝害敏感(waterlogging-sensitive, W-S)基因型大豆冀豆17为材料, 采用荧光定量PCR、Illumina MiSeq高通量测序技术, 分析了不同淹水时间下2个基因型根际细菌多样性、群落组成和网络特征。结果表明, 耐涝基因型大豆的生物量和细菌丰度明显高于涝害敏感基因型大豆。主坐标分析(PCoA)表明, 耐涝基因型与敏感基因型大豆微生物群落组成的差异随淹水时间的增加而变化(< 0.05)。在淹水条件下, 耐涝基因型大豆富集了属和属以及OTU274 ()和OTU2334 ()等物种, 这些细菌的富集可能与耐涝性有关, 本研究提供了大豆根际微生物抗涝潜力的证据。
大豆; 耐涝; 根际微生物; 16S rRNA; 网络分析
未来极端气候事件的发生频率将会大幅度增加,其中包含了干旱、暴雨和随之而来的涝灾[1-2]。在美国、加拿大、巴西、法国、中国和日本等一些粮食生产关键地区受到极端气候事件的影响尤为显著[3-5]。大豆(L.)是对涝害适应性较差的作物, 当大豆遭受涝害时, 土壤中的氧气含量非常低, 造成了根部呼吸作用减弱进而导致严重的营养供给危机[6-8]。此外, 涝害胁迫还会干扰作物的生理功能, 包括叶片光合作用减少、叶、茎和根的气孔封闭以及生长抑制, 从而导致产量下降[9-10]。因此, 我们应该尽可能多的理解关于作物耐涝的机制, 包括土壤微生物在缓解涝害中的作用[11-12]。
目前已有多种方法用于提高作物对涝害胁迫的适应能力, 包括传统的植物育种、作物的基因工程和植物相关微生物组的管理和应用[13-14]。作物的植物育种和基因工程可以帮助植物更好地适应胁迫环境[15]。然而, 许多耐胁迫基因型的研究并未考虑土壤环境的生物和非生物因素, 特别是微生物对植物的胁迫响应所产生的耐性作用[16-18]。已有研究表明, 根系相关微生物不仅直接受到涝害的影响, 同时也间接受到植物对胁迫的响应的影响[19]。作物受到涝害胁迫, 导致地下碳输入数量及质量发生变化[9,20], 进而影响了根际微生物组[21]。不同植物种类对土壤过程和反馈的特征存在差异, 且不同植物基因型在根际会招募不同的微生物组, 因此土壤微生物群落在介导植物对涝害胁迫的反应中发挥重要作用[22-23]。
近年来, 越来越多的研究关注于植物微生物组介导的抵抗胁迫研究[24]。例如, 含1-氨基环丙烷-1-羧酸脱氨酶的有益细菌可以降低胁迫诱导的乙烯含量[25-26], 进而保护植物免受淹水[27]、干旱[28]和高盐[29]等胁迫的有害影响。耐性品种可以通过募集特定的微生物来抵抗外界胁迫[30-31]。我们基于不同耐铝大豆根际微生物对铝毒的响应机制研究发现, 耐铝大豆会招募耐铝的细菌和, 可以有效帮助耐铝大豆减轻Al的毒性[31]。然而关于大豆涝灾的研究, 多数集中关注遗传和栽培措施的改进, 而关于不同基因型大豆的根际微生物如何响应涝害的相关研究还未见报道。
基于此, 本研究选取耐涝基因型大豆齐黄34和涝害敏感基因型大豆冀豆17为材料, 通过评估不同淹水时长下耐涝和涝害敏感基因型大豆根际细菌多样性及群落结构, 来解析涝害程度对不同大豆品种根际微生物的影响差异。本研究强调了根际细菌群落作为潜在育种目标的可能性, 以生产对涝害胁迫更耐受的作物。
1 材料与方法
1.1 供试材料
1.1.1 供试土壤 供试土壤为酸性土, 采自广东省英德市(113°42′E, 24°28′N), 其包含全碳10.30 g kg-1、全氮0.33 g kg-1、全钾12.15 g kg-1、速效氮143.5 mg kg-1、速效磷1.33 mg kg-1、速效钾115.9 mg kg-1, pH 4.75。
1.1.2 供试大豆 盆栽试验供试大豆(L.)包括涝害敏感型大豆: W-S (冀豆17-JD17)[32]; 耐涝大豆: W-T (齐黄34-QH34)[33]。冀豆17由河北省农林科学院粮油作物研究所张孟臣研究员提供, 齐黄34由山东省农业科学院作物研究所徐冉研究员提供。
1.2 大豆土培盆栽试验
本试验在广东省广州市华南农业大学农学院进行, 采用土培盆栽的方式, 设置2个大豆品种(JD17和QH34), 1个无淹水对照, 2个淹水时长处理, 共6个处理, 每个处理设置6个重复。2个淹水时长为1 d和5 d。试验前将风干土壤过2 mm的筛, 去除杂质备用。采用规格相同的花盆(外径13.8 cm, 内径13.0 cm, 底径10.4 cm, 高12.2 cm), 每盆装风干土约2.5 kg。在播种前2~3 d, 保持土壤含水量为田间持水量的50%~60%。播种时, 每盆播8粒外形一致、壮实饱满的大豆种子, 生长过程在可控条件的玻璃房中进行(白天温度为28~32℃, 夜间温度为16~20℃), 出苗6 d后定苗至3株大豆, 出苗期再移至自然光线良好充足的室外继续生长, 期间按时管理和除草。生长期间的土壤含水量控制在田间持水量的80%左右。
播种大豆后待其生长至花期, 进行淹水处理。取与种植用花盆等量的相同规格的花盆作为隔水用花盆。将塑料膜裁剪成合适大小的方片, 紧贴着隔水用花盆内壁覆盖花盆, 用以阻隔水分, 防止水分从花盆底下流失, 保证淹水处理效果。塑料膜铺好后需要高出花盆1~2 cm。确认无破孔漏洞后, 在种植用花盆外能直接套入外层隔水用花盆, 两花盆之间能紧密贴合。添加水量至淹没土壤表面以上2 cm, 每日2次定时定量加水, 观察并记录生长情况, 分别于水淹后1 d、5 d时戳破花盆低漏洞处的塑料膜恢复排水, 观察并记录。
1.3 土壤样品的收集
不同处理的大豆分别在淹水后1 d和5 d采用“抖根法”采集根际土壤[34]。试验总共6个处理, 每个处理设置6个重复, 共采集36个样品。由于淹水胁迫下, 土壤含水量高于最大田间持水量, 土壤黏结成块状, 实际操作时, 需要托起沥去部分水分, 轻轻撇掉与大豆根系结合松散的土壤, 然后取下与大豆根系紧密附着的土壤作为根际土壤样品, 保存于−80℃冰箱。随后将所有土壤样品送深圳华大基因科技服务有限公司, 完成土壤微生物总DNA的提取、PCR扩增以及测序数据的初步处理。
1.4 DNA提取和qPCR扩增
利用Fast kit DNA试剂盒提取根际土壤总DNA, 提取的DNA在去离子水中稀释, 并储存于-20℃冰箱中保存备用。使用具有12 nt barcode引物338F (5′-ACTCCTACGGGAGGCAGCAG-3′)和806R (5′- GGACTACHVGGGTWTCTAAT-3′)扩增16S rRNA基因的V3–V4高变区[35]。qPCR反应体系包括22.5 μL的PCR SuperMix、1.0 μL正向引物和1.0 μL反向引物、10 ng模板DNA, ddH2O补充至25 μL。PCR扩增体系95℃变性10 min; 95℃ 15 s, 60℃ 10 s, 72℃ 10 s, 28个循环; 72℃延伸10 min[36]。
1.5 数据分析
使用QIIME v1.19.1处理原始序列数据[37]。删除小于200 bp和平均质量分数低于20的序列[38]。通过运行UCHIME算法检测潜在的嵌合序列[39]。用CD-HIT按97%相似性对序列进行OTU聚类[40]。使用RDP对数据库执行OTU代表序列的分类归属[41-42]。所有样本均随机重采样至相同序列深度(每个样本29,169个序列), 以减少测序深度对处理效果的影响。在QIIME v1.19.1中计算根际细菌Alpha-多样性(Chao1和Shannon指数)。并且基于Bray-Curtis距离矩阵进行主坐标分析(PCoA)用于分析不同处理之间微生物群落组成的相似性[43]。利用非参数多元方差分析(Adonis)计算微生物群落结构的差异。采用检验(Student’s-test)对相对丰度在前100的属进行差异性分析, 并利用Duncan’s法进行多重比较。Venn分析用于统计不同处理样本中所共有和独有的细菌属的数量。采用GenStat统计软件进行大豆生物量、细菌丰度和多样性指数的单因素方差分析(ANOVA), 并利用Duncan’s法来进行多重比较, 显著性水平均设为0.05。
利用共现网络评价相对丰度大于0.2%的细菌OTUs之间的关系。利用R包psych[44]计算Spearman相关系数, 得到OTU之间的成对相关, 网络中包含> 0.8和< 0.05的相关。利用Gephi v.0.9.2为每个植物基因型和淹水处理构建了共现网络可视化[45-46]。计算网络的拓扑性质, 以阐明不同基因型和淹水处理的群落结构差异。
2 结果与分析
2.1 大豆生物量、根际细菌丰度及多样性
2种基因型在不淹水和淹水1 d处理中生物量无显著差异, 在淹水5 d时, 耐涝基因型大豆生物量显著高于涝害敏感大豆(图1-a)。淹水降低了大豆根际微生物细菌的基因拷贝数, 淹水5 d处理中耐涝基因型的细菌基因拷贝数明显高于敏感基因型(图1-b,<0.05)。由不同淹水处理根际土壤细菌的多样性指数(图1-c, d)表明, 淹水处理没有对2个基因型大豆根际微生物多样性产生影响。
2.2 不同处理根际细菌的群落结构
对36个土壤样品测序共获得了1,304,142条有效序列, 每个样品19,852~37,180条, 平均35,603条, 在97%相似度下聚类得到5538个OTU。在6个处理中只检测到41个门, 其中变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿湾菌门(Chloroflexi)、浮霉菌门(Planctomycetes)和疣微菌门(Verrucomicrobia)为优势菌门, 其相对丰度分别为31.0%~33.7%、14.8%~17.7%、4.6%~8.7%、7.0%~ 8.3%、6.1%~7.0% (图2)。随着淹水时长的增加, 耐涝基因型大豆根际中的变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)的相对丰度增加, 但是绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)、疣微菌门(Verrucomicrobia)的相对丰度随着淹水时间的增加而减少。随着淹水时长的增加, 涝害敏感基因型大豆中的酸杆菌门(Acidobacteria)相对丰度随着淹水时长的增加而增加, 浮霉菌门(Planctomycetes)的相对丰度随淹水时间增加而减少(图2)。在属水平上, 在无淹水处理、淹水1 d和淹水5 d处理中, 耐涝和敏感基因型大豆根际分别有9、13和12个属显著不同。在无淹水对照处理中, 有5个属的相对丰度在耐涝基因型大豆中更高(图3-a, b)。然而, 在淹水1 d和5 d处理下, 各有4个属的相对丰度在耐涝基因型中更高, 其中和在2个处理中的耐涝害基因型中共同增高(图3-c)。
为评价不同涝害处理下耐涝和敏感基因型大豆OTU相对丰度的差异, 建立了负二项分布的广义线性模型。结果表明, 相对于耐涝不淹水处理, 耐涝淹水处理1 d和5 d的根际土壤样本中显著富集的OTU分别有150个和290个(图4-a, b); 相对于敏感不淹水处理, 敏感淹水1 d和5 d处理的根际土壤样本中显著富集的OTU分别有100个和230个(图4-c, d)。通过Venn分析表明, 18个OTU仅在淹水1 d和5 d处理中富集在耐涝基因型大豆根际土壤中(图4-e)。其中OTU218 ()、OTU938 ()、OTU274 ()、OTU2334 ()、OTU1337 ()分别在2个大豆基因型淹水处理下存在显著差异, 淹水5 d处理中, OTU274 ()、OTU2334 ()在耐涝基因型中的相对丰度较高(图5)。
不同字母表示相同处理下2个品种间差异显著(< 0.05)。W-TCK; 耐涝品种淹水0 d; W-SCK: 涝害敏感品种淹水0 d; W-T1D: 耐涝品种淹水1 d; W-S1D: 涝害敏感品种淹水1 d; W-T5D: 耐涝品种淹水5 d; W-S5D: 涝害敏感品种淹水5 d。
Boxes superscripted by different letters indicate significant differences between two varieties under the same treatment (< 0.05). W-TCK: waterlogging-tolerant variety without waterlogging; W-SCK: waterlogging-sensitive variety without waterlogging; W-T1D: waterlogging-tolerant variety under waterlogging for one day; W-S1D: waterlogging-sensitive variety under waterlogging for one day; W-T5D: waterlogging-tolerant variety under waterlogging for five days; W-S5D: waterlogging-sensitive variety under waterlogging for five days.
处理同图1。Treatments are the same as those given in Fig. 1.
误差条表示6个重复样本的标准差。采用Benjamini-Hochberg法校正值(< 0.05)。处理同图1。
The error bars show the calculated standard variation of six replicates. Corrected-values were calculated by the Benjamini-Hochberg false discovery rate approach at (< 0.05). Treatments are the same as those given in Fig. 1.
a: 耐涝品种淹水1 d和不淹水的对比分析; b: 耐涝品种淹水5 d和不淹水的对比分析; c: 涝害敏感品种淹水1 d和不淹水的对比分析; d: 涝害敏感品种淹水5 d和不淹水的对比分析; e: 不同淹水处理下根际土壤细菌在OTU水平的Venn分析。处理同图1。
a: comparison analysis of W-T variety under waterlogging for one day and no waterlogging; b: comparison analysis of W-T variety under waterlogging for five days and no waterlogging; c: comparison analysis of W-S variety under waterlogging for one day and no waterlogging; d: comparison analysis of W-S variety under waterlogging for five days and no waterlogging; e: Venn analysis of rhizosphere soil bacteria at genus level under different waterlogging time. Treatments are the same as those given in Fig. 1.
W-T: 耐涝基因型大豆; W-S: 涝害敏感基因型大豆。不同字母表示相同处理下2个品种间差异显著(< 0.05)。处理同图1。
W-T: waterlog-tolerant soybean genotypes; W-S: waterlog-sensitive soybean genotypes. Bars superscripted with different letters indicate significant differences between the two varieties under the same treatment at< 0.05. Treatments are the same as those given in Fig. 1.
2.3 根际土壤细菌群落的结构
PCoA结果显示(图6), X和Y轴共解释了65.2%的群落变异, 并且6个处理分成3个大组, 其中不同淹水处理的根际细菌群落聚集在一起, 说明淹水处理是改变根际微生物群落结构的主要因素。此外, 不同处理下2种基因型的根际细菌群落结构呈现显著差异, 而且这个差异随着淹水时长的增加也发生了变化(图6和表1)。
处理同图1。Treatments are the same as those given in Fig. 1.

表1 通过变异多变量方差分析(PERMANOVA)评估大豆基因型对根际细菌群落结构的影响
处理同图1。**表示1%显著水平;*表示5%显著水平。
Treatments are the same as those given in Fig. 1.**Significant at the 1% probability level;*Significant at the 5% probability level.
2.4 细菌群落的关联网络分析
网络关联分析表明, 在不同淹水处理下, 耐涝和敏感基因型大豆根际土壤之间的网络结构存在显著差异(图7和表2)。随着涝害时间的增加, 耐涝基因型的正相关数、图密度和平均加权度增加, 而在敏感基因型中网络的正相关数在淹水处理下更高。随着淹水时长的增加, 耐涝和敏感基因型的平均加权度在淹水1 d和5 d处理下分别比不淹水有所增高。淹水5 d处理下, 耐涝基因型的正相关和负相关链接数都高于涝害敏感基因型。随着淹水时长的增加, 耐涝基因型网络变得更复杂, 值得注意的是敏感基因型中的网络也变得比不淹水时复杂。
3 讨论
3.1 不同淹水时长对根际土壤微生物多样性的影响
本研究旨在揭示涝害胁迫对耐涝基因型大豆和涝害敏感基因型大豆根际细菌的影响。当大豆遭受长时间淹水处理后, 细菌丰度都出现降低趋势, 且耐涝基因型高于涝害敏感型。该结果与Azarbad等[47]的研究结果一致, 他们发现涝害急速降低了小麦根际土壤细菌的丰度。然而该结果与郭太忠等[48]的研究结果相反, 他们发现涝害增加了玉米根际细菌的数量。造成这一不同的原因可能有2个: (1) 2个研究利用的方法不同, 郭太忠等[48]利用的是平板计数法, 而该研究利用的是荧光定量PCR的方法。(2) 作物种类的不同可能也会造成相反的结果。该研究中, 耐涝品种的根际拥有更多的细菌数量, 这也与Azarbad等[47]的研究结果相似, 涝害对不同作物基因型的根际细菌影响差异显著。这可能是因为土壤在淹水条件下, 氧气大量减少, 好氧细菌不易存活[49-50], 此外, 淹水胁迫下耐涝基因型和敏感基因型根部不定根数量、通气组织强弱程度、根瘤数量、电导率和丙二醛含量等参数也可能造成耐涝品种拥有更多的细菌数量[47,51-54]。最后, 涝害胁迫强烈影响大豆植物的初级和次级代谢, 且大多数改变的化合物都参与碳和氮的代谢以及苯丙烷途径, 且这些改变在耐涝品种和涝害敏感品种中的差异是不一样的,其可能也是造成耐涝品种拥有更多的细菌数量的重要原因之一[55]。
a: 耐涝品种淹水0 d时根际细菌网络结构; b: 涝害敏感品种淹水0 d时根际细菌网络结构; c: 耐涝品种淹水1 d时根际细菌网络结构; d: 涝害敏感品种淹水1 d时根际细菌网络结构; e: 耐涝品种淹水5 d时根际细菌网络结构; f: 涝害敏感品种淹水5 d时根际细菌网络结构。图上不同的颜色节点表示不同的门。红色连接线表示2个节点正相关, 蓝色连接线表示2个节点负相关。处理同图1。
a: co-occurrence network of the rhizosphere bacterial community of W-T variety without waterlogging; b: co-occurrence network of the rhizosphere bacterial community of W-S variety without waterlogging; c: co-occurrence network of the rhizosphere bacterial community of W-T variety under waterlogging for one day; d: co-occurrence network of the rhizosphere bacterial community of W-S variety under waterlogging for one day; e: co-occurrence network of the rhizosphere bacterial community of W-T variety under waterlogging for five days; f: co-occurrence network of the rhizosphere bacterial community of W-S variety under waterlogging for five days. Different color nodes represent different phyla. The red connection line indicates positive correlation between two nodes, and the blue connection line indicates negative correlation between two nodes. Treatments are the same as those given in Fig. 1.
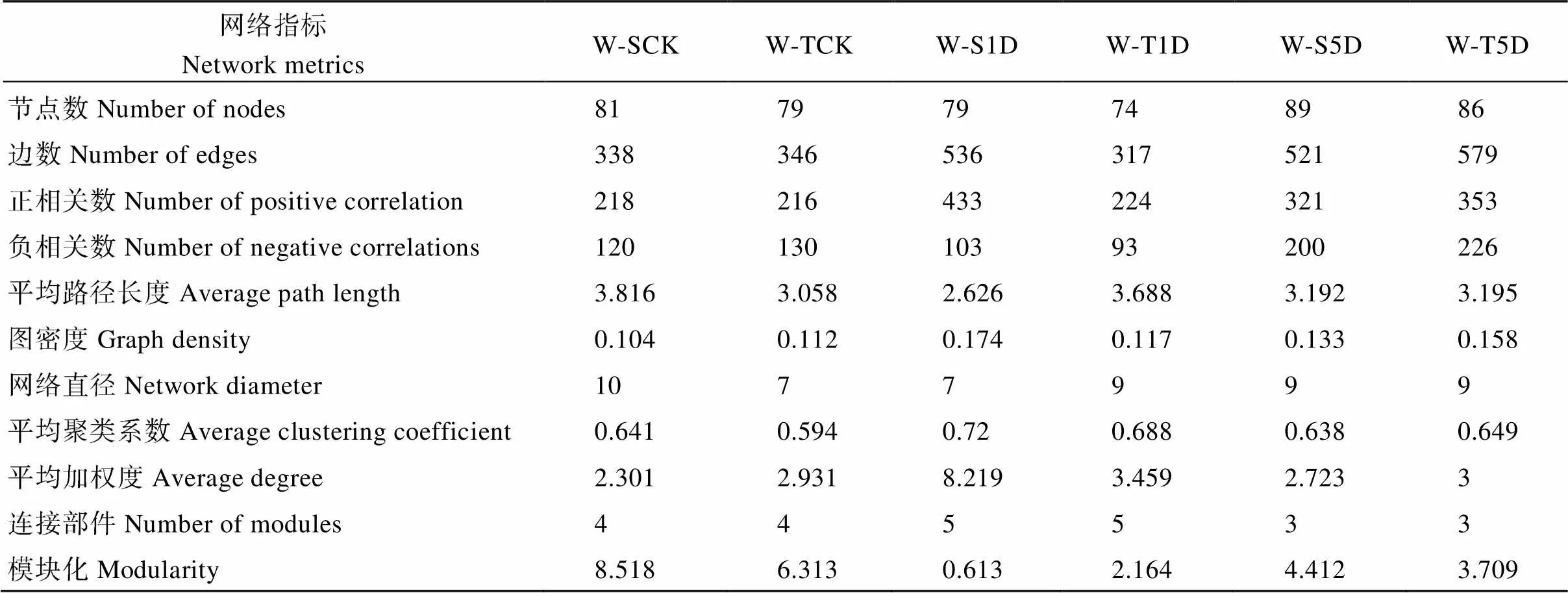
表2 根际细菌网络的拓扑结构
处理同图1。Treatments are the same as those given in Fig. 1.
3.2 不同淹水时长对根际土壤微生物群落结构的影响
基于PCoA分析结果发现, 涝害显著影响了大豆根际细菌群落结构(图6)。这与前人研究的结果一致, 涝害会对作物的根际土壤微生物群落结构产生影响[56]。涝害可以对根际微生物造成直接的影响, 当涝害导致厌氧时, 对环境极为敏感的土壤微生物呼吸速率和活动减少, 进而群落结构发生变化[57-62]。此外, 作物对涝害胁迫的响应也间接影响着根际微生物群落结构[12]。已有研究表明, 当作物受到涝害胁迫会影响地下碳输入的变化[19-20], 进而影响了根际微生物组[63]。
本研究还发现, 涝害显著改变了不同大豆基因型的根际微生物群落结构(表1)。这是因为不同的植物基因型响应涝害胁迫的机制是不同的, 比如, 与涝害敏感大豆相比, 耐涝大豆在淹水后, 其根系生长、根系损伤以及气孔的速率表现出更高的适应性[9,48]。此外, 淹水对大豆植株根部的一级代谢和二级代谢均有较大影响。大多数改变的化合物涉及碳和氮代谢以及苯丙醇途径, 而涝害敏感大豆和耐涝大豆之间的反应是不同的[64]。以上的不同都会对根际微生物产生直接的影响, 造成了涝害条件下2个品种根际微生物群落结构的显著差异。
基于属水平上的分析我们发现, 在淹水时长为1 d和5 d时, 耐涝基因型的细菌属和显著高于敏感基因型。其中, 细菌属属于放线菌门, 而前人研究表明, 放线菌能够定殖在植物上促进根瘤细菌的分化, 还能够促进土壤中的铁离子的吸收, 增加植物抗性[65-66]。属于浮酶状菌纲, 浮酶状菌纲的一些属可以在厌氧条件下处于厌氧氨氧化过程, 而且还参与碳循环[67-69]。因此, 上述和在耐涝处理下相对丰度的增加, 说明耐涝品种可能通过促进作物对碳氮元素及微量离子元素的吸收, 进而增加大豆对涝害的抵抗力。然而, 在2个基因型中差异表达OTU与耐涝性的相关是通过统计方式实现的, 因此其在耐涝过程中具体的功能和作用还需要进一步进行验证。
基于OTU水平上的分析发现, 淹水处理5 d时, 耐涝基因型大豆富集了OTU274 ()和OTU2334 () (图5)。是厌氧异养促生菌, 这些属的一些种具有可以定植在根系周围产生丙酸、分解纤维素的功能, 可能会帮助植物获取更多的营养物质适应胁迫[70-71]。有研究报道在土壤中的硫酸盐还原和碳循环中起着关键作用[72], 然而该物种在耐涝害中的作用还没有报道。
3.3 不同淹水时长对不同大豆基因型细菌关联网络的影响
在农田环境中, 微生物物种是不可能单独生存的, 而是与其他自然界物种形成复杂的生态网络[73]。网络分析的结构特性不仅可以揭示物种之间的复杂关系, 还可以表征生态网络结构的稳定性[62,74]。本研究进行了关联网络分析, 以获得对细菌群落组成的更全面了解, 随着耐涝基因型的淹水时长的增加, 正相关数、图密度和平均加权度增加, 表明耐涝基因型的网络被复杂化, 而敏感基因型中观察到淹水处理也使得网络较为复杂。但是在水淹5 d处理下, 耐涝基因型的网络比敏感基因型更加复杂化, 耐涝基因型的正相关数、负相关数、平均度都高于涝害敏感基因型。这和我们的前期研究结果相似, 即非生物胁迫下, 抗性品种的网络结构更为复杂[75]。前人研究表明, 高度连接的微生物群可以激发植物免疫系统, 加速激活对外界的防御[76-78]。表明涝害使耐涝基因型的网络复杂化, 从而提高大豆抵御涝害胁迫的能力[78-79]。
4 结论
不同淹水处理对不同基因型大豆的土壤根际细菌群落产生影响。随着淹水时长的增加, 2个基因型大豆的细菌丰度逐渐降低, 且在淹水时长为5 d的时候也出现了显著差异, 耐涝基因型的细菌丰度高于涝害敏感型基因的细菌丰度。耐涝基因型大豆在淹水处理到达5 d后富集了属和属以及OTU274 ()和OTU2334 (), 这些物种的富集可能与耐涝相关, 但其在耐涝过程中具体的功能和作用还需要进一步进行验证。此外, 涝害胁迫复杂化耐涝基因型大豆的网络结构, 这可能提高土壤细菌群落抵御其他生物和非生物因素胁迫的能力。
[1] Loreti E, Van V H, Perata P. Plant responses to flooding stress., 2016, 33: 64–71.
[2] Duggan B L, Domitruk D R, Fowler D B. Yield component variation in winter grown under drought stress., 2000, 80: 739–745.
[3] Bagci S A, Ekiz H, Yilmaz A, Cakmak I. Effects of zinc deficiency and drought on grain yield of field-grown wheat cultivars in Central Anatolia., 2007, 193: 198–206.
[4] Jiang D, Fan X, Dai T, Cao W. Nitrogen fertilizer rate and post anthesis waterlogging effects on carbohydrate and nitrogen dynamics in wheat., 2008, 304: 301–314.
[5] Yang M, Wangr F. Effects of tea and fungus intercropping on soil microbial community of tea., 2010, 11: 13–16.
[6] Fukao T, Bailey S J. Submergence tolerance conferred byis mediated by SLR1 and SLRL1 restriction of gibberellin responses in rice., 2008, 105: 16814– 16819.
[7] Setter T L, Waters I. Review of prospects for germplasm improvement for waterlogging tolerance in wheat barley and oats., 2003, 253: 1–34.
[8] Herzog M, Striker G G, Colmer T D, Pedersen O. Mechanisms of waterlogging tolerance in wheat: a review of root and shoot physiology., 2016, 39: 1068–1086.
[9] Grayston S, Wang J, Campbell C D S, Edwards A C. Selective influence of plant species on microbial diversity in the rhizosphere., 1998, 30: 369–378.
[10] Sairam R K, Dharmar K, Chinnusamy V, Meena R C. Waterlogging-induced increase in sugar mobilization, fermentation, and related gene expression in the roots of mung bean ()., 2009, 166: 602–616.
[11] Ngumbi E, Kloepper J. Bacterial-mediated drought tolerance: current and future prospects., 2016, 105: 109–125.
[12] Nguyen L T T, Osanai Y, Lai K, Anderson I C, Bange M P, Tissue D T, Singh B K. Responses of the soil microbial community to nitrogen fertilizer regimes and historical exposure to extreme weather events: flooding or prolonged-drought., 2018, 118: 227–236.
[13] Fleury D, Jefffferies S, Kuchel H, Langridge P. Genetic and genomic tools to improve drought tolerance in wheat., 2010, 61: 3211–3222.
[14] Quiza L, St-Arnaud M, Yergeau E. Harnessing phytomicrobiome signaling for rhizosphere microbiome engineering., 2015, 6: 507–516.
[15] Coleman D D, Tringe S G. Building the crops of tomorrow: advantages of symbiont-based approaches to improving abiotic stress tolerance., 2014, 5: 283–295.
[16] Budak H, Akpinar B A, Unver T, Turktas M. Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC-ESI-MS/MS., 2013, 83: 89–103.
[17] Swamy B P M, Kumar A. Genomics-based precision breeding approaches to improve drought tolerance in rice., 2013, 31: 1308–1318.
[18] Waterer D, Benning N T, Wu G, Luo X, Liu X, Gusta M. Evaluation of abiotic stress tolerance of genetically modified potatoes (cv. Desiree)., 2010, 25: 527–540.
[19] Sanaullah M, Blagodatskaya E, Chabbi A, Rumpel C, Kuzyakov Y. Drought effects on microbial biomass and enzyme activities in the rhizosphere of grasses depend on plant community composition., 2011, 48: 38–44.
[20] Canarini A, Dijkstra F. Dry-rewetting cycles regulate wheat carbon rhizodeposition, stabilization and nitrogen cycling., 2015, 81: 195–203.
[21] Grayston S J, Wang S, Campbell C D, Edwards A C. Selective influence of plant species on microbial diversity in the rhizosphere., 1998, 30: 369–378.
[22] Lau J A, Lennon J T. Rapid responses of soil microorganisms improve plant fitness in novel environments., 2012, 35: 14058–14062.
[23] Kaisermann A, Vries F T, Grifths R I, Bardgett R D. Legacy effects of drought on plant-soil feedbacks and plant-plant interactions., 2017, 215: 1413–1424.
[24] Castrillo G, Teilxeira P, Paredes S, Theresa F L, Laura L, Meghan E, Feltcher O M, Finkel N W, Breakfield P M, Corbin D J, Javier P A, Jeffery L. Root microbiota drive direct integration of phosphate stress and immunity., 2017, 543: 513–518.
[25] Saleem M, Arshad M, Hussain S, Bhatti A S. Perspective of plant growth promoting rhizobacteria (PGPR) containing ACC deaminase in stress agriculture., 2007, 34: 635–648.
[26] Barnawal D, Bharti N, Maji D, Chanotiya C S, Kalra A. 1-Aminocyclopropane-1-carboxylic acid (ACC) deaminase-containing rhizobacteria protectplants during waterlogging stress via reduced ethylene generation., 2012, 58: 227–235.
[27] Grichko V P, Glick B R. Flooding tolerance of transgenic tomato plants expressing the bacterial enzyme ACC deaminase controlled by the 35S,or PRB-1promoter.2001, 39: 19–25.
[28] Zahir Z A, Munir A, Asghar H N, Shaharoona B, M Arshad. Effectiveness of rhizobacteria containing ACC-deaminase for growth promotion of pea () under drought conditions., 2007, 18: 958–963.
[29] Mayak S, Tirosh T, Glick B R. Plant growth-promoting bacteria confer resistance in tomato plants to salt stress., 2004, 42: 565–572.
[30] Lebeis S L, Paredes S H, Lundberg D S, Breakfield N, Gehring J, McDonald M, Malfatti S T, Tijana G, Corbin D, Susannah G, Jeffery L. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa., 2015, 349: 860–864.
[31] Lian T X, Ma Q B, Shi Q H, Cai Z D, Zhang Y F, Cheng Y B, Nian H. High aluminum stress drives different rhizosphere soil enzyme activities and bacterial community structure between aluminum-tolerant and aluminum-sensitive soybean genotypes., 2019, 440: 409–425.
[32] 赵青松, 闫龙, 刘兵强, 邸锐, 史晓蕾, 赵双进, 张孟臣, 杨春燕. 高产广适优质大豆品种冀豆17. 大豆科学, 2015, 34: 736–739.
Zhao Q S, Yan L, Liu B Q, Di R, Shi X L, Zhao S J, Zhang M C, Yang C Y. High yield wide adaptability and high quality soybean variety Jidou 17., 2015, 34: 736–739 (in Chinese with English abstract).
[33] 徐冉, 王彩洁, 张礼凤, 李伟, 戴海英, 张军. 高产优质多抗广适大豆新品种齐黄34的选育. 山东农业科学, 2013, 45(3): 107–108.
Xu R, Wang C J, Zhang L F, Li W, Dai H Y, Zhang J. Breeding of a new soybean variety Qihuang 34 with high yield, high quality and multi resistance., 2013, 45(3): 107–108 (in Chinese with English abstract).
[34] Castrillo G, Teilxeira P, Paredes S, Law T, Lorenzo L, Feltcher M, Finkel O, Breakfield N, Mieczkowski P, Jones C, Paz J, Dangl J. Root microbiota drive direct integration of phosphate stress and immunity., 2017, 543: 513–518.
[35] Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Noah F, Antonio G P, Julia K G, Jeffrey I G, Gavin A H, Scott T K, Dan K, Jeremy E K. QIIME allows analysis of high-throughput community sequencing data., 2010, 7: 335–336.
[36] Magoc T, Salzberg S L. FLASH: fast length adjustment of short reads to improve genome assemblies., 2011, 27: 2957–2963.
[37] Edgar R, Haas B, Clemente J, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection., 2011, 27: 2194–2200.
[38] Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences., 2006, 22: 1658–1659.
[39] Abarenkov K, Nilsson R H, Larsson K H, Alexander I J, Eberhardt U, Erland S, Klaus H, Rasmus K, Ellen L, Taina P, Robin S, Andy F S, Taylor L, Björn M U, Trude V. The UNITE database for molecular identification of fungi recent updates and future perspectives., 2010, 186: 281–285.
[40] Wang Q, Garrity G M, Tiedje J M, Cole J R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy., 2007, 73: 5261–5267.
[41] Muyzer G, Waal E D, Uitterlinden A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction amplified genes coding for 16S rRNA., 1993, 59: 695–700.
[42] Li J, Lin J, Pei C, Kaitao L, Thomas C J, Guang D T. Variation of soil bacterial communities along a chronosequence of Eucalyptus plantation., 2018, 6: 5648–5657.
[43] Revelle W. Procedures for personality and psychological research., 2017, 7: 136–142.
[44] Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks., 2009, 361–362.
[45] Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks., 2014, 5: 219–232.
[46] Agler M T, Ruhe J, Kroll S, Morhenn C, Kim S T, Weigel D, Eric M K. Microbial hub taxa link host and abiotic factors to plant microbiome variation., 2016, 14: e1002352.
[47] AzarbadH, Constant P, GiardL C, Bainard L D, Yergeau E. Water stress history and wheat genotype modulate rhizosphere microbial response to drought., 2018, 126: 228–236.
[48] 郭太忠, 袁刘正, 赵月强, 柳家友, 谷川. 渍涝对玉米产量和根际土壤微生物的影响. 湖北农业科学, 2014, 53: 505–507.
Guo T Z, Yuan L Z, Zhao Y Q, Liu J Y, Gu C. Effects of waterlogging on maize yield and the rhizosphere soil microorganism., 2014, 53: 505–507 (in Chinese with English abstract).
[49] 曾成城, 陈锦平, 魏虹, 刘媛, 马文超, 王婷, 周翠. 水淹生境下秋华柳对Cd污染土壤微生物数量及酶活性的影响. 生态学报, 2017, 37: 4327–4334.
Zeng C C, Chen J P, Wei H, Liu Y, Mao W C, Wang T, Zhou C. Effects ofon soil microorganisms and enzymatic activity in contaminated soils under flooding conditions., 2017, 37: 4327–4334 (in Chinese with English abstract).
[50] 赵可夫. 植物对水涝胁迫的适应. 生物学通报, 2003, 38(12): 11–14.
Zhao K F. Adaptation of plants to waterlogging stress., 2003, 38(12): 11–14 (in Chinese with English abstract).
[51] Went F W. Effect of root system on tomato stem growth., 1943, 18: 51–65.
[52] 倪君蒂, 李振国. 淹水对大豆生长的影响. 大豆科学, 2000, 19: 42–48.
Ni J D, Li Z G. Effects of flooding on soybean growth., 2000, 19: 42–48 (in Chinese).
[53] 宋晓慧, 滕占林, 箫长亮, 李冬梅, 李文滨, 张代平. 淹水胁迫对不同耐涝性大豆品种苗期根部形态及叶部生理指标的影响. 大豆科学, 2014, 32: 130–132.
Song X H, Teng Z L, Xiao C L, Li D M, Li W B, Zhang D P. Effect of waterlogging on root morphology and foliar physiological indexes of soybean varieties., 2014, 32: 130–132 (in Chinese with English abstract).
[54] Constant P, Chowdhury S P, Hesse L, Pratscher J, Conrad R. Genome data mining and soil survey for the novel group 5 [NiFe]-hydrogenase to explore the diversity and ecological importance of presumptive high affinity H2-oxidizing bacteria., 2011, 77: 6027–6035.
[55] Duarte C I, Mertz H L M, Aurelian D S, Nepomuceno A, Moraes L, Alexandra C, Marcolino G J, Richter C, Colnago L A. Flooded soybean metabolomic analysis reveals important primary and secondary metabolites involved in the hypoxia stress response and tolerance., 2018, 153: 176–187.
[56] Greening C, Biswas A, Carere C R, Jackson C J, Taylor M C, Stott M B, Cook G M, Morales S E. Genomic and metagenomic surveys of hydrogenase distribution indicate H is a widely utilised energy source for microbial growth and survival., 2016, 10: 761–777.
[57] Evans S E, Wallenstein M D. Soil microbial community response to drying and rewetting stress: does historical precipitation regime matter., 2012, 109: 101–116.
[58] Preece C, Peñuelas J. Rhizodeposition under drought and consequences for soil communities and ecosystem resilience., 2016, 409: 1–17.
[59] Unger I M, Kennedy A C, Muzika R M. Flooding effects on soil microbial communities., 2009, 42: 1–8.
[60] Fierer N, Schimel J, Holden P. Variations in microbial community composition through two soil depth profiles., 2003, 35: 167–176.
[61] Moyano F E, Manzoni S, Chenu C. Responses of soil heterotrophic respiration to moisture availability: an exploration of processes and models., 2013, 59: 72–85.
[62] Meisner A, Leizeaga A, Rousk J, Bååth E. Partial drying accelerates bacterial growth recovery to rewetting., 2017, 112: 269–276.
[63] Fuchslueger L, Bahn M, Fritz K, Hasibeder R, Richter A. Experimental drought reduces the transfer of recently fixed plant carbon to soil microbes and alters the bacterial community composition in a mountain meadow., 2014, 201: 916–927.
[64] Kozlowski T T. Flooding and plant growth., 1994, 91: 107.
[65] Coutinho I D, Baker J M, Ward J L, Beale M H, Creste S, Cavalheiro A. Metabolite profiling of sugarcane genotypes and identification of flavonoid glycosides and phenolic acids., 2016, 64: 4198–4206.
[66] Tokala R K, Strap J L, Jung C M, Jung D L, Crawford M S, Lee A, Deobald J, Franklin B. Novel plant-microbe rhizosphere interaction involvingWYEC108 and the pea plant ()., 2002, 68: 2161–2171.
[67] Yamanaka K, Oikawa H, Ogawa H, Hideaki T, Shohei S, Teruhiko B, Kenji U. Desferrioxamine E produced bystimulates growth and development of., 2005, 151: 2899–2905.
[68] 黄佩蓓, 焦念志, 冯浩, 舒青龙. 海洋浮霉状菌多样性与生态学功能研究进展. 微生物学报, 2014, 41: 1891–1902.
Huang P B, Jiao N Z, Feng H, Shu Q L. Research progress on Planctomycetes’ diversity and ecological function in marine environments., 2014, 41: 1891–1902 (in Chinese with English abstract).
[69] Gloeckner F O, Bauer M, Teeling H, Lombardot T, Ludwig W, Gade D, Beck A, Borzym K, Heitmann K, Rabus R, Schlesner H, Amann R, Reinhardt R. Complete genome sequence of the marine planctomycetesp. Strain 1., 2003, 103: 292–310.
[70] Strobel G. Harnessing endophytes for industrial microbiology., 2006, 9: 240–244.
[71] Somers E, Vanderleyden J, Srinivasan M. Rhizosphere bacterial signalling: a love parade beneath our feet., 2004, 30: 205–240.
[72] Zhang Y H P, Lynd L R. Cellulose utilization by Clostridium thermocellum: bioenergetics and hydrolysis product assimilation., 2005, 56: 168–176.
[73] Pester M, Bittner N, Deevong P, Wagner M, Loy A. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland., 2010, 4: 1591–1602.
[74] Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E. The large-scale organization of the bacterial network of ecological co-occurrence interactions., 2010, 38: 3857–3868.
[75] Shi Q, Liu Y, Shi A, Cai Z, Nian H, Martin H, Lian T. Rhizosphere soil fungal communities of aluminum-tolerant and -sensitive soybean genotypes respond differently to aluminum stress in an acid soil., 2020, 11: 1177.
[76] Xiao X, Liang Y, Zhou S, Zhuang S, Sun B. Fungal community reveals less dispersal limitation and potentially more connected network than that of bacteria in bamboo forest soils., 2018, 27: 550–563.
[77] Jones J D G, Dang J L. The plant immune system., 2006, 444: 323–329.
[78] Peter N D, John P R. Plant immunity: towards an integrated view of plant-pathogen interactions., 2010, 11: 539–548.
[79] Van der Ent S, Van Hulten M, Pozo M J, Czechowski T, Udvardi M K, Pieterse C M J, Ton J. Priming of plant innate immunity by rhizobacteria and β-aminobutyric acid: differences and similarities in regulation., 2009, 183: 419–431.
Effects of waterlogging on rhizosphere microorganisms communities of different soybean varieties
YU Tao-Bing, SHI Qi-Han, NIAN-Hai*, and LIAN Teng-Xiang*
College of Agriculture, South China Agricultural University / Guangdong Subcenter of National Soybean Improvement Center, Guangzhou 510642,Guangdong, China
Waterlogging affects the composition of rhizosphere microbial community of different soybean varieties. The tolerance of soybean plant with different genotypes to waterlogging is quite different. In this study, waterlogging tolerant soybean genotype (Qihuang 34) and waterlogging sensitive soybean genotype (Jidou 17) were selected. The bacterial diversity, community composition, and network characteristics in the rhizosphere of the two genotypes under different waterlogging time were analyzed via fluorescence quantitative qPCR and Illumina Miseq high-throughput sequencing. The results showed that the biomass of waterlogging tolerant genotype and bacterial abundance in its rhizosphere were significantly higher than those for waterlogging sensitive genotype. The PCoA analysis showed that the difference in microbial community composition between waterlogging tolerant and sensitive soybean genotypes changed with waterlogging time (< 0.05). Under the condition of waterlogging,and, OTU274 () and OTU2334 () enriched in the rhizosphere of the waterlogging tolerant genotype. The enrichment of these bacteria might be related to waterlogging tolerance. This study provides evidence of the microbial potential in the rhizosphere of soybean against waterlogging.
soybean; waterlogging tolerance; rhizosphere microorganism; 16S rRNA; network analysis
10.3724/SP.J.1006.2021.04137
本研究由国家重点研发计划项目“大田经济作物优质丰产的生理基础与调控”(2018YFD1000900)资助。
This study was supported by the National Key Research and Development Program of China “Physiological Basis and Agronomic Management for High-quality and High-yield of Field Cash Crops” (2018YFD1000900).
连腾祥, E-mail: liantx@scau.edu.cn; 年海, E-mail: hnian@scau.edu.cn
E-mail: 277885643@qq.com
2020-06-23;
2020-09-13;
2020-09-22.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200922.1143.006.html
