An improved draft genome sequence of hybrid Populus alba × Populus glandulosa
2021-07-15XiongHuangSongChenXiaopengPengEunKyungBaeXinrenDaiGuimingLiuGuanzhengQuJaeHeungKoHyoshinLeeSuChenQuanziLiMengzhuLu
Xiong Huang·Song Chen·Xiaopeng Peng·Eun-Kyung Bae·Xinren Dai·Guiming Liu·Guanzheng Qu·Jae-Heung Ko·Hyoshin Lee·Su Chen·Quanzi Li·Mengzhu Lu
Abstract Populus alba × P.glandulosa clone 84 K,derived from South Korea,is widely cultivated in China and used as a model in the molecular research of woody plants because of high gene transformation efficiency.Here,we combined 63-fold coverage Illumina short reads and 126-fold coverage PacBio long reads to assemble the genome.Due to the high heterozygosity level at 2.1% estimated by k-mer analysis,we exploited TrioCanu for genome assembly.The PacBio clean subreads of P.alba × P.glandulosa were separated into two parts according to the similarities,compared with the parental genomes of P.alba and P.glandulosa.The two parts of the subreads were assembled to two sets of subgenomes comprising subgenome A (405.31 Mb,from P.alba)and subgenome G (376.05 Mb,from P.glandulosa) with the contig N50 size of 5.43 Mb and 2.15 Mb,respectively.A high-quality P.alba × P.glandulosa genome assembly was obtained.The genome size was 781.36 Mb with the contig N50 size of 3.66 Mb and the longest contig was 19.47 Mb.In addition,a total of 176.95 Mb (43.7%),152.37 Mb (40.5%)of repetitive elements were identified and a total of 38,701 and 38,449 protein-coding genes were predicted in subgenomes A and G,respectively.For functional annotation,96.98% of subgenome A and 96.96% of subgenome G genes were annotated with public databases.This de novo assembled genome will facilitate systematic and comprehensive study,such as multi-omics analysis,in the model tree P.alba × P.glandulosa.
Keywords Genome assembly·Gene annotation·Hybrid poplar·Populus alba × p.glandulosa cl.84 K
Introduction
Forests play important roles in maintaining ecological equilibrium on earth.Populus(poplar) is one of the major tree genera cultivated widely for both ecological and economic objectives.The genus has been used as a model in research because of rapid growth,small genome,and successful transformation system (Bradshaw et al.2000).Currently,system biology with multi-omics integration has been applied to poplar to identify key factors controlling important traits and to construct genetic regulator networks,especially for wood formation (Yuan et al.2008;Moreno-Risueno et al.2010;Li et al.2014).The efficient integration of genomics and other omics relies on the quality of genome sequences.Since first completion of genome sequencing of the speciesP.trichocarpa(Torr.and Gray) (Tuskan et al.2006),high quality genome sequences have been assembled for white poplar,P.albaL.and its var.phramidalis(Liu et al.2019;Ma et al.2019) and in other two speciesP.euphraticaOliv.andP.pruinosaSchrenk that grow in deserts (Ma et al.2013;Yang et al.2017).Although a transformation system inP.trichocarpahas been established (Song et al.2006;Li et al.2017),it is still not successful in some laboratories.This species can only grow in some regions,limiting its spread for research.Hybrid poplar,P.alba×P.glandulosacl.84 K,widely cultivated in China and Korea,is used as a model to study tree growth and disease resistance (Zhang et al.2011;Ke et al.2017;Yoon et al.2018).A high transformation efficiency has been achieved using leaves as explants (Li et al.2017),attracting more research laboratories to use this hybrid.Moreover,the improved genome sequence ofP.alba×P.glandulosawill provide important reference for future research and it will facilitate systematic and comprehensive studies,such as multi-omics analysis,molecular breeding practices,and comparative genomic analysis with other species and hybrids.
Materials and methods
Plant materials,DNA and RNA extraction
Fresh young leaves and stem differentiating xylem were collected from a single 1-year-oldP.alba×P.glandulosaplant in the YuQuanShan garden of the Chinese Academy of Forestry in Beijing and immediately frozen in liquid nitrogen.Young leaves ofP.glandulosawere also sampled for DNA extraction using the modified CTAB (hexadecyl trimethyl ammonium bromide) method (Porebski et al.1997).In addition,tissues of leaves,stem xylem and phloem ofP.alba×P.glandulosawere collected from three individual trees and used for total RNA isolation using the CTAB method (Chang et al.1993).
Library construction and sequencing
Both xylem and leaf DNA from the hybrid were used for PacBio (Pacific Biosciences,Memlo Park,CA,USA) singlemolecule real-time (SMRT) sequencing because the xylem tissues contain fewer chloroplasts compared to leaves.Libraries were constructed using~ 20 kilobases (kb) size DNA fragments following the manual of PacBio.A total of 58 SMRT cells were sequenced,of which 50 from xylem DNA libraries were run on the PacBio RS II platform;six cells from xylem DNA libraries and two cells from leaf DNA libraries were run on the PacBio Sequel platform.A total of 59.72 gigabases (Gb) subreads with an N50 length of 8151 base pair (bp) were generated.After removing adaptors,correcting sequences and selecting the subreads of more than 5000 bp,46.67 Gb of clean subreads were obtained and used for the de novo genome assembly (Table S1).
In addition,three Illumina-shorted DNA libraries with an insert size of~ 350 bp were constructed forP.alba×P.glandulosausing the TruSeq DNA Sample Prep Kit (Illumina Inc.,San Diego,CA,USA).Libraries were pair-end sequenced with 150 bp both sides on the Illumina HiSeq X-10 platform,resulting in a total of 30 Gb raw data.The raw reads were subjected to a quality check and filtered by fastp (https://githu b.com/OpenG ene/fastp).The raw reads were processed by removing adapter sequences,low-quality reads and possible contaminated reads.In total,27.6 Gb of clean data were obtained (Table S1).An Illumina-shorted DNA library with insert size of~ 550 bp was constructed forP.glandulosausing Truseq Nano DNA Prep Kit.The library was pair-end sequenced with 151 bp both sides on the Illumina Novaseq 6000 platform,resulting in a total of 46.5 Gb raw data (Table S2).
Transcriptome sequencing
After quality checking on an Agilent Bioanalyzer 2100,total RNA of leaves,phloem and xylem,each with three biological replicates from three individual trees,were used for RNA-seq library construction using the NEB Next Ultra Directional RNA Library Prep Kit for Illumina (Ispawich,NE,USA) following the manufacturer’s instructions.The nine libraries were sequenced using the Illumina HiSeq 4000 platform with a read length of 150 bp at both sides.Raw data with 25.6 Gb,23.9 Gb and 25.7 Gb were obtained for leaves,phloem and xylem,respectively.More than 57 million paired-end reads were generated for each sample(Table S3).These RNA-seq reads were then assembled using Trinity (Grabherr et al.2011) with the default parameters,and a total of 103,140 (> 300 bp) fragments were obtained for subsequent transcriptome-based gene prediction.All sequencing reads for genome and transcriptome have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject number PRJNA526157.
Estimation of genome size and heterozygosity
To guide genome assembly,the genome size ofP.alba×P.glandulosawas determined by 17-mer frequency distribution analysis with KMC (Marek et al.2017) and GenomeScope(Vurture et al.2017) using the 27.6 Gb Illumina sequencing reads.The final result was plotted as a frequency graph(Fig.1).The genome size was estimated to be 474.43 Mb.The two distinctive peaks observed from the distribution curve demonstrate a high heterozygosity of theP.alba×P.glandulosagenome,with an estimated heterozygosity level of 2.1% (Table S4),indicating a 97.9% sequence identity between parental haplotypes.
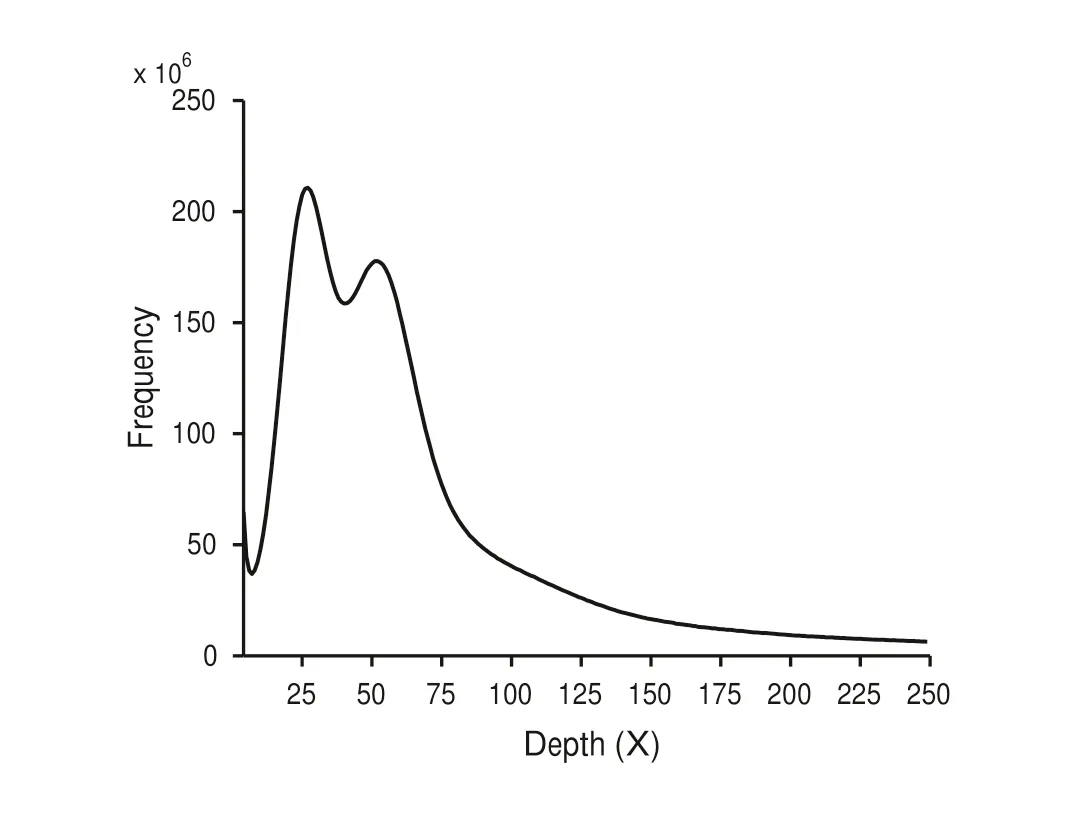
Fig.1 17-mer analysis for P.alba × P.glandulosa genome;x-axis is the k-mer depth,the y-axis is the frequency of the k-mer for a given depth
Genome assembly
The higher heterozygosity level increases the difficulty of genome assembly.Xu et al.(2020) used Canu (Koren et al.2017) to assemble the genome ofGardenia jasminoidesJ.Ellis,which had a comparable level of heterozygosity asP.alba×P.glandulosa.We conducted genome assembly using the TrioCanu (Koren et al.2018) software,a newly developed de novo sequence assembler especially for the assembly of haplotype-resolved sequences from diploid genomes.The genome sequence ofP.alba(Liu et al.2019) was downloaded from NCBI (Accession number:PRJNA491245) and used as a template to simulate 45 Gb Illumina reads for haplotype phasing by pirs (https ://githu b.com/galax y001/pirs).We also obtained 46.5 Gb Illumina reads forP.glandulosa.The Illumina sequencing reads from the two parental genomes were used to partition the long reads from an offspring into haplotype-specific sets,and each haplotype was assembled independently,resulting in a complete diploid reconstruction.The separated PacBio reads were corrected,trimmed and assembled into contigs using Canu v2.0 (Koren et al.2017) with the default parameters.The resultant contigs were polished with Racon (Vaser et al.2017) independently.
Repeat annotation
Repetitive sequences and transposable elements (TEs) in the two sets of subgenomes (A and G) were respectively identified and classified using a combination of de novo and homology-based approaches at DNA level.Initially,a de novo repeat library using RepeatModeler (Tarailo-Graovac and Chen 2009) was built with default parameters.RepeatMasker (https://www.repea tmask er.org/) was then conducted to map the assemblies against both the databases that we had constructed and the known Repbase(Jurka et al.2005) TE library.In this process,the Dfam(Hubley et al.2016) database version is 3.0 and the Repbase version is 20,181,026.
Gene annotation
We exploited MAKER3 pipeline (Cantarel et al.2008)which required sufficient evidence from a combination of protein,EST and ab initio gene prediction input.For homology-based prediction,annotated proteins from five sequenced plants (P.trichocarpa,Ricinus communis,Arabidopsis thaliana,Salix suchowensisandVitis vinifera) were independently mapped to the subgenomes A and G using TBLASTN (Camacho et al.2009) with default parameters.TheP.alba×P.glandulosatranscriptome fragments based on Inchworm assembly were used as the EST evidence in MAKER3 pipeline.For ab initio gene prediction,the results from the above RepeatMasker analysis were used as the reference in the repeat sequences analysis,with eukaryotic selected in the organism type.We adopted the three methods SNAP (Korf 2004),GeneMark(Alexandre et al.2005) and Augustus (Stanke and Waack 2003) for de novo annotation for the repetitive sequences masked genome.
Gene function annotation was performed with several protein databases (SwissProt,KEGG,BIGG,and COG) to predict protein domains for functional information.In addition,Gene Ontology (GO) annotation was performed by AHRD (https://githu b.com/group schoo f/AHRD),which is an informative select description and GO terms tool.SwissProt (Brigitte et al.2003) was selected as the database for protein queries,and the weights were assigned by default.three tissues were matched to theP.alba×P.glandulosa

Comparative genome analysis with P.trichocarpa
The number of protein-coding genes and the results of BUSCO analysis with the same parameters were compared betweenP.alba×P.glandulosagenome andP.trichocarpav4.1 genome.Gene similarity analysis was also carried out to compare protein-coding genes between these two.We independently conducted genome structure analysis between subgenome A andP.trichocarpa,and between subgenome G andP.trichocarpausing MCScanX software (Wang et al.2012) and genome sequences as inputs.Some monolignol biosynthetic pathway enzyme genes in subgenomes A and G were identified through BioEdit (Alzohairy 2011),and their gene structures were compared with paralogous genes inP.trichocarpausing the GSDS 2.0 pipeline (Hu et al.2015).
Results
Genome assembly and assessment
Consequently,two sets of subgenomes were assembled,comprising subgenome A (405.31 Mb fromP.alba) and subgenome G (376.05 Mb fromP.glandulosa) with the contig N50 size of 5.43 Mb and 2.15 Mb,respectively.By merging the sequences of two subgenomes together,we obtained a high-qualityP.alba×P.glandulosagenome assembly with a total length of 781.36 Mb containing 2,109 contigs(N50=3.66 Mb;N90=168.21 Kb) and the longest contig was 19.47 Mb (Table 1).The distribution of the average guanylic and cytidylic acid (GC) content of theP.alba×P.glandulosagenome was 34.1%,similar to that inP.euphratica(32.1%) (Ma et al.2013) andP.trichocarpa(33.6%)(Tuskan et al.2006).
Three methods were carried out to evaluate the accuracy and the completeness of theP.alba×P.glandulosagenome,subgenomes A as well as G.A total of 27.6 Gb (~ 59X)Illumina short reads were aligned back to theP.alba×P.glandulosagenome,subgenome A and subgenome G using BWA (Md et al.2019) with default parameters and the analysis showed that 99.4%,98.7% and 97.8% of the reads were successfully mapped,respectively.A BUSCO (Simão et al.2015) and MUMmer (Kurtz et al.2004) analyses were conducted.BUSCO analysis with the recently released plant dataset from OrthoDB (https ://www.ortho db.org) showed that 98.1%,98.5%,96.9% (1349,1354,1332 out of 1375)of the core eukaryotic genes had been recovered in theP.alba×P.glandulosagenome,subgenome A and subgenome G,respectively.A total of 2,344,256 SNPs sites and 1,460,279 short INDELs were identified,and the divergence between the parental haplotypes was estimated to be 3% with MUMmer.The RNA-seq reads generated in this study from genome,subgenome A and subgenome G using BWA (Li 2013),showing a more than 98.0% mapping rate (Table S3).All assessments above indicated that theP.alba×P.glandulosagenome assembly was qualified for the subsequent applications.
Repeat and gene annotation
In total,the annotation indicated approximately 43.7%(176.95 Mb) repeat sequences for subgenome A,and 40.5% (152.37 Mb) for subgenome G.Of which,long terminal repeats (LTRs) were the most abundant repeat class,accounting for 49.1% and 46.9% of repetitive sequences,representing 21.4% and 19.0% of the subgenomes A and G,respectively (Table S5).Tandem repeats were also identified using Tandem Repeat Finder (TRF) package (Gary 1999),and the results showed a total of 169,369 and 152,982 repeats in subgenomes A and G,respectively.
After removing the unreasonable genes through the revision function of MAKER3,a total of 38,701 and 38,449 protein-coding genes in the subgenomes A and G with an average coding sequence (CDS) length of 1,122.86 and 1,112.34 bp,respectively were annotated (Table 2).To assess the completeness of the two sets of subgenomes,the BUSCO analysis with the recently released plant dataset from OrthoDB was executed (Simão et al.2015).The results showed that 95.0%,93.5% (1306,1286 out of 1375)of the core eukaryotic genes could be presented in subgenome A and G,respectively.In addition,37,532 (97.0%)protein-coding genes in subgenome A and 37,280 (97.0%)in subgenome G can be annotated in at least one database.After the BLASTP comparison,a total of 6,979,765 and 6,924,093 hits occurred and in the process of summarizing the information for the genes,GO annotation information was obtained for 37,286 and 37,051 genes,accounting for 96.3%,96.4% of the total number of genes for subgenomes A and G,respectively (Table 2).
Comparative genome analysis with P.trichocarpa
Coding genes were annotated in theP.alba×P.glandulosagenome and the low-quality genes were removed.A total of 77,150 (subgenome A:38,701;G:38,449) coding genes were identified for theP.alba×P.glandulosagenome,comparable to the gene number (34,699) inP.trichocarpav4.1 genome.The gene numbers inPopulusgenomes are generally higher than in other genomes with a median size(400–500 Mb).ThePopulusgenus underwent a recent whole genome duplication event,and nearly 8000 pairs of paralogous genes of similar age,(excluding tandem or local duplications),were generated after polyploidization.Moreover,the current annotation of theP.alba×P.glandulosagenome covered 1,333 (97.0%) of the 1,375 BUSCOs and 16 (1.2%)are fragmented.The completeness of the annotated genes inP.trichocarpausing BUSCOs was assessed with the same parameters.Version 4.1 annotation ofP.trichocarpacovered 1,372 (99.8%) of the 1,375 BUSCOs without fragments.
SinceP.trichocarpais the first poplar species with genome sequencing and its genome has been well annotated,the protein-coding genes inP.alba×P.glandulosawere compared with those inP.trichocarpa.A total of 61,929(subgenome A:31,271;G:30,658) protein-coding genes,accounting for 80.3% in theP.alba×P.glandulosagenome,have high similarities with the genes inP.trichocarpa.Of which,45,442 (subgenome A:22,933;G:22,509) genes have > 90.0% sequence similarity with their paralogous genes inP.trichocarpaand 33,894 (subgenome A:17,147;G:16,747) genes have > 95.0% sequence similarity with their paralogous genes inP.trichocarpa.Synteny analysis on genome structure showed that 257 blocks existed between subgenome A andP.trichocarpaand 253 blocks between subgenome G andP.trichocarpa(Fig.2).We identified 28 genes encoding key enzymes in monolignol biosynthetic pathway in subgenomes A and G (Table 3).Three genes with corresponding symbol names were regarded as a group.Three genes in one group had the same number of exons andintrons.The lengths of three genes in most groups looked similar,such asPAL1,CCoAOMT1,C4H2,CAld5H2groups.While the lengths of three genes in several groups exhibited obvious differences,the length of intron inPtrCSE1was longer than inPag_A_CSE1andPag_G_CSE1inCSE1group,and the length of intron inPtrCAld5H1was shorter than inPag_A_CAld5H1andPag_G_CAld5H1inCAld5H1group (Fig.3).Whether the variations in length cause differences in enzyme activities needs further investigations.
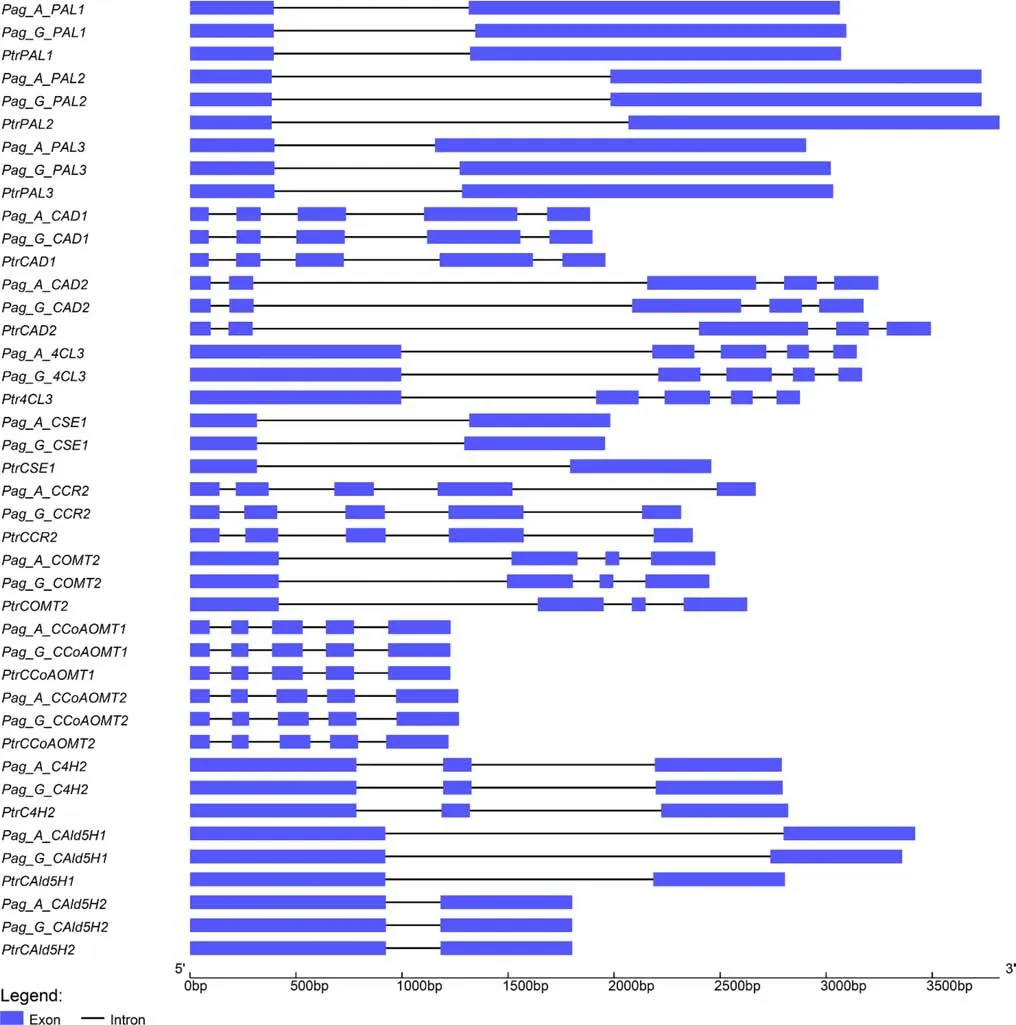
Fig.3 Structure analysis for paralogous genes in subgenome A,subgenome G and P.trichocarpa;genes encode enzymes in monolignol biosynthetic pathway

Table 3 Statistics of paralogous genes identified in subgenomes A and G

Fig.2 Synteny analysis of genome structure between subgenome A and P.trichocarpa,and between subgenome G and P.trichocarpa independently;sequences with lengths more than 1 Mb were selected in subgenomes A and G
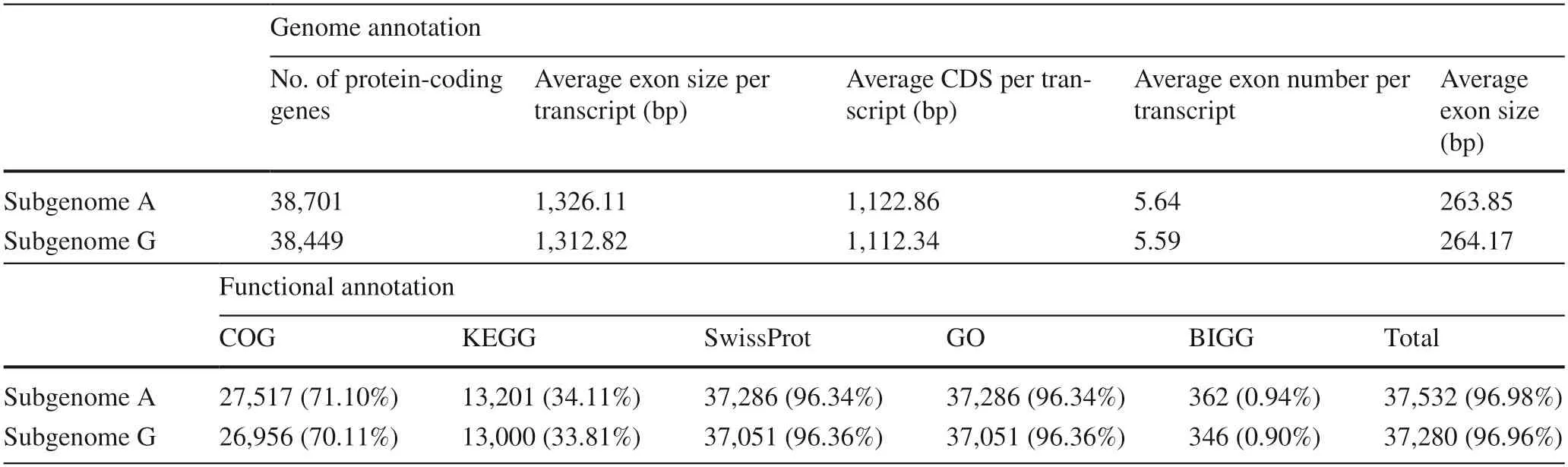
Table 2 Summary of genome annotation and functional annotation of P.alba × P.glandulosa comprising subgenomes A and G
Discussion
De novo assembler selection
Nowadays,more and more genomes of plant species have been sequenced.At the same time,many bioinformatic tools for genome de novo assembly were exploited,such as Canu(Koren et al.2017),Smartdenovo (Istace et al.2017) and Wtdbg2 (Ruan and Li 2019).Due to different genome ploidy and high genome heterozygosity,complex allelic variation hampers the assembly of haplotype-resolved sequences from diploid genomes.It is crucial to choose a suitable software for high quality genome assembly.Each genome assembly strategy and software used have advantages and limitations.For example,Wtdbg2 is a newly developed de novo sequence assembler for long noisy reads produced by PacBio or Oxford Nanopore Technologies.It assembles raw reads without error corrections and usually collapses heterozygosity.ALLHIC (Zhang et al.2019) has been used for genome assembly in autopolyploid sugarcaneSaccharum spontaneumL.It is a new Hi-C scaffolding pipeline,specifically tailored to the highly heterozygous diploids or polyploid genomes.The advent of long-read sequencing technologies has not overcome the challenge of completely assembling both haplotypes of a diploid genome.Instead,most genome assembly tools simply co-assemble the haplotypes into a mosaic consensus,resulting in an assembly that does not accurately represent either original haplotype.Collapsing haplotypes into a single consensus representation introduces false variants that are not present in either haplotype,leading to annotation and analysis errors (Korlach et al.2017).Ideally,a genome should be represented as a complete set of haplotypes rather than an artificial mixture.TrioCanu (Koren et al.2018) uses short reads from two parental genomes to first partition long reads from an offspring into haplotype-specific sets,simplifying haplotype assembly by resolving allelic variation prior to assembly.Each haplotype is then assembled independently,resulting in a complete diploid reconstruction.Here,to better avoid cross interference of genome information from parental sequences and improve assembly accuracy,we chose TrioCanu (Koren et al.2018) to assemble the genome of hybrid diploidP.alba×P.glandulosa,which has a high heterozygosity.This software has been used in the genome assembly ofBos taurusandB.taurus indicusand demonstrate that the quality of each haplotype exceeds that of even the best livestock reference genomes (Koren et al.2018).Choosing a suitable software is vital for genome assembly and further influences genome annotation.
Genome assembly and annotation quality
High-quality genome assembly and annotation provide foundation for accurate downstream analysis.In this study,we assembledP.alba×P.glandulosagenome of 781.36 Mb,comprising two sets of subgenomes A and G.Compared with the genome assembly by Qiu et al.(2019),the contig N50 size of our assembly (3.66 Mb) was 1.6 times bigger at contig level.We used several methods in the gene annotation.By integrating the results from different approaches,we obtained sufficient evidence to make annotation result more accurate and complete.The BUSCO evaluations of genome assembly and annotation both showed a > 97.0% coverage of the 1375 BUSCOs,and near 97.0% protein-coding genes in either subgenome A or subgenome G were functionally annotated.The protein-coding genes inP.alba×P.glandulosawere compared with those inP.trichocarpaand 80.3%genes had similarities with the genes inP.trichocarpa,exhibiting a comparative annotation level.In total,our assembly and annotation ofP.alba×P.glandulosagenome can satisfy the following functional genomic analysis.
Conclusions
P.alba×P.glandulosais a model hybrid to study growth,wood formation and stress response.The flowering ofP.alba×P.glandulosahas been surveyed in several locations in China over 10 years and it was found that some trees are monoecious,providing an ideal material to study sex determination in poplar.The improved assembly ofP.alba×P.glandulosais noted and the comparison of the genomes betweenP.alba×P.glandulosaandP.trichocarpashows that this assembled genome can accelerate the study of tree growth and response to adverse environments,improving productivity and resistance to stress.The genome assembly and annotation files are available at Figshare database (https://doi.org/10.6084/m9.figsh are.12369 209).
杂志排行

Journal of Forestry Research的其它文章
- Flexible transparent wood enabled by epoxy resin and ethylene glycol diglycidyl ether
- Diversity and surge in abundance of native parasitoid communities prior to the onset of Torymus sinensis on the Asian chestnut gall wasp (Dryocosmus kuriphilus) in Slovenia,Croatia and Hungary
- Ozone disrupts the communication between plants and insects in urban and suburban areas:an updated insight on plant volatiles
- Testing visible ozone injury within aLight Exposed Sampling Site as aproxy for ozone risk assessment for European forests
- Logging and topographic effects on tree community structure and habitat associations in a tropical upland evergreen forest,Ghana
- Spatial pattern dynamics among co-dominant populations in early secondary forests in Southwest China