同源DNA序列比对缺失位点的核苷酸最大似然插补
2021-07-13潘克迈秦雪瑞刘雄恩
潘克迈, 秦雪瑞, 刘雄恩
(福建农林大学计算机与信息学院,福建 福州 350002)
分子系统发育推算中采用的DNA进化模型如JC69、K80、F81、F84、HKY85、TN93、REV94等,均为描述4种核苷酸之间变换的马尔可夫随机过程[1,2],用于分子进化分析的同源DNA多序列比对数据中出现的比对缺失(gap)成为上述模型应用的障碍.目前通用的数据预处理方法是将含gap位点成对或整列删除.DNA序列比对数据代表了初始的同源性假设,忽略gap的数据处理方式会丢失分子进化信息,造成系统发育树枝长估算的偏差和进化距离的偏低估计[3-5].因此,在分子系统发育分析推算中融合gap信息是必要的.
Mcguire et al[6]将gap视作核苷酸的第5种状态,首次提出了融合gap信息的JC69+gap、F81+gap、F84+gap等5-状态模型,基于模型进化距离的估算仍会出现偏差.林碧娇等[7]在上述3种模型基础上引入新的参数,提出JC69+gap′、F81+gap′、F84+gap′等改进模型,区分了indel与substitution在性质上的差异.基于改进模型推导的核苷酸变换概率5阶矩阵可以应用于系统发育重建方法中的最大似然法.改进模型F81+gap′减小了进化距离估算的偏差,但其中F84+gap′模型的参数较多、计算复杂且未能推导出相应的距离计算公式[8].目前流行的系统发育重建软件,如PHYLIP[9]、PAUP[10]、MEGA[11]、PAML[12]、MrBayes[13]、PhyML[14]等,未将上述5-状态模型作为计算模型的参考选项.
秦雪瑞等[8]将序列比对后出现的gap视为统计抽样过程中产生的随机缺失数据,提出对同源DNA多序列比对数据中的gap进行核苷酸插补等数据预处理方法;给出了核苷酸最近邻插补算法,并比较分析插补前后序列基于4-状态模型及比对序列基于5-状态模型的序列间进化距离;对核苷酸最近邻插补的数据预处理方法进行了评估.
本文基于最大似然法进一步提出DNA序列gap核苷酸最大似然插补算法,并比对缺失位点删除、核苷酸最近邻插补以及本文提出的最大似然插补等3种数据预处理方法在进化距离估算、系统发育重建等方面的效果,旨在提高DNA分子系统发育重建树枝长估算的精确度.
1 方法和原理
1.1 最大似然估计的一般方法
最大似然法是一种参数估计的统计学方法,它利用总体的相关概率密度函数以及样本所提供的信息来求解未知参数的估计量.其原理是:从总体中随机抽取n个样本单元,使样本观测值出现的可能(概率密度函数似然值)达到最大的参数估计量是最合理的[15].
若离散型总体X的分布函数为P{X=x}=p(x;θ),θ∈Θ,其中θ是一个未知参数或几个未知参数组成的参数向量,Θ是θ的可能取值范围.设X1、X2、 …、Xn是来自X的样本,x1、x2、…、xn是对应于样本X1、X2、…、Xn的样本观测值.已知样本X1、X2、 …、Xn取到观察值x1、x2、…、xn的概率,则事件{X1=x1,X2=x2,…,Xn=xn}发生的概率为:
(1)
这一概率随θ的取值而变化,L(θ)称为样本的似然函数.最大似然估计法固定样本观察值x1,x2,…,xn,在θ取值的可能范围(Θ)筛选使似然函数L(x1,x2,…,xn;θ)达到最大参数值,作为参数θ的估计值.即
(2)
为了优化计算,实际应用中通常以对数似然函数的最大值来估算θ,即
(3)
1.2 比对缺失位点的核苷酸最大似然插补算法
基于最小进化原理[16],本文假设同源DNA比对序列在核苷酸插补前后采用4-状态DNA进化马尔可夫模型,与运用最大似然法推算的进化树拓扑一致,即二者系统发育重建树的差异仅在于枝长的估计值.基于这个假设,首先在插补前将gap位点删除后选择4-状态模型,运用上述系统发育重建的最大似然法建立系统发育树拓扑,并估算枝长和模型的其它参数;再通过使单个位点似然函数值达到最大的方法估算gap位点最大可能的核苷酸状态.
以图1所示的3个同源序列系统发育树拓扑为例,3个序列在一特定位点上的核苷酸状态观察数据分别为A、-、C,分枝结点为0和4.连接到第j个结点的枝长tj定义为平均每个位点核苷酸置换的期望数,在重建系统发育树拓扑时已临时估算.估算结点2序列在该缺失位点上最大似然核苷酸的似然函数:
(4)
式中,πx0是结点0序列核苷酸的频率.核苷酸插补的最大似然法需要估计的参数是gap位点的核苷酸θ,θ=x,x∈{A,T,C,G}.
同源DNA序列中gap位点核苷酸最大似然插补算法描述如下:
Algorithm Nucleotide Interpolation by MLI// maximum likelihood interpolation
Begin
Input multi-aligned DNA sequencesX
GetX′ by deleting all sites with gaps inX
Infer phylogenetic treeT by maximum likelihood withX′
Fors← 1stTo the last gap site inXDo
Begin
k← number of sequences with gap ats
Fori← 1stTo the 4k-th combination of nucleotide for k gaps atsDo
ComputeLiunderT, likelihood value for thei-th combination
IfLh= max (Li) ThenX1..k,s← theh-th combination of nucleotide,X1..k,s∈{T,C,A,G}
Forj← 2ndTo the last sequence Do
IfXj,s≠X1,sThen break Else continue loop
Ifj> count of sequences Then delete sitesElse remains
End
OutputXafter nucleotide interpolation at gap sites
End
与最近邻插补法一样,插补后,若各序列在该位点的核苷酸完全相同,则删除该位点(整列),否则保留插补后位点.剔除插补后核苷酸相同的位点,因为插补后这种位点在分子进化分析中不提供进化信息,而且会减低序列间进化距离的估算精度[8].
对于长度为n含有s个gap位点的m个多序列比对数据矩阵Xm×n,每个gap位点所在列最多存在m-1个gap,则核苷酸最大似然法中插补算法的时间复杂度为O(4m-1s).
2 结果与分析
2.1 测试序列
本文选取3组文献采用的同源DNA序列[8],用ClustalX进行多序列比对后,分别采取比对缺失位点整列删除(Del)、核苷酸最近邻插补(MNI)和核苷酸最大似然插补(MLI)等3种数据预处理,估算序列间进化距离;并用MEGA中的最大似然法在K80模型下进行500次系统发育重建的测试试验.
利用计算机重复60次模拟,结果如图2所示.系统发育树拓扑的DNA进化过程,其中一组仅有substitution事件,另一组包含indel事件.DNA分子进化的计算机模拟是基于分子钟假说[17,18],采取固定时序随机抽取位点和沿树进化序列的方法[19].模拟过程记录真实树各分枝枝长以及8个同源DNA终端序列.30组仅有substitution事件的DNA多序列比对数据不含gap,在无需数据预处理的情形下重建系统发育树,以重建树枝长与真实树枝长的误差作为基准误差;另30组记录包含indel事件的同源序列分别在3种预处理下重建树的枝长;并分析不同预处理方法在系统发育重建时的误差.

图1 系统发育树拓扑某位点示意图Fig.1 A phylogenetic tree of three homologous DNA sequences at a site
2.2 测试结果
2.2.1 不同预处理方法的进化距离 表1、2、3分别显示第1、2、3组文献数据[8]在Del、MNI和MLI数据预处理下序列间进化距离的估算结果.

表1 猿类7个物种线粒体DNA序列在不同预处理和模型下的进化距离Table 1 Evolutionary distances of mitochondrial DNA sequences of 7 apes under different preprocessing and models

表2 睡莲科6种植物核糖体DNA中ITS在不同预处理和模型下的进化距离Table 2 Evolutionary distances of rDNA ITS2 region of 6 Nymphaeaceae plants under different preprocessing and models
2.2.2 不同预处理方法下的系统发育重建 图3、4、5分别显示第1、2、3组文献数据[8]在Del、NNI和MLI等3种数据预处理下系统发育重建的结果(分别位于图的上、中、下部分).
从表1~3可知:比对缺失位点的核苷酸插补,无论是MNI算法还是MLI算法预处理后的序列间进化距离都大于Del预处理下的距离估算.在同源多序列比对序列矩阵中直接删除含有gap位点的列,突变信息丢失是造成其序列间进化距离偏低估计的原因.
从图3~5可知:MNI和MLI核苷酸插补的预处理方法下系统发育重建的进化树拓扑与Del预处理下重建进化树的拓扑是一致的,自展检验结果具有很高且几乎一致的置信度.其次,证实了核苷酸插补前后基于4-状态DNA进化马尔可夫模型的系统发育重建进化树具有相同的拓扑结构,差异仅在于枝长的估计值.
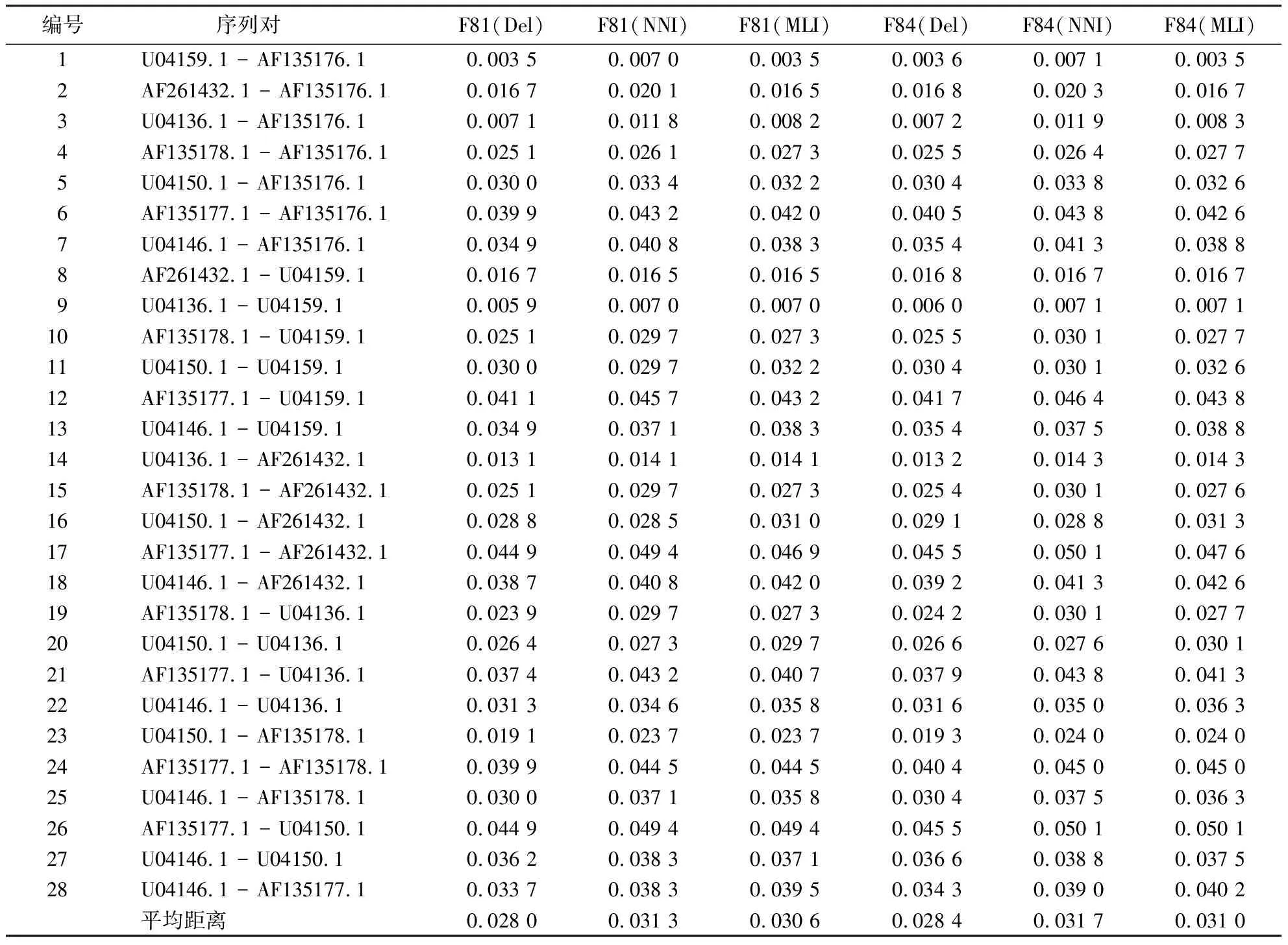
表3 侧耳属8种真菌25S rRNA序列在不同预处理和模型下的进化距离Table 3 Evolutionary distances of 25S rRNA sequences of 8 Pleurotus fungus under different preprocessing and models

图3 第1组数据在3种预处理后重建的系统发育树Fig.3 Reconstructed phylogenetic trees from processed data of group 1 by 3 preprocessing methods
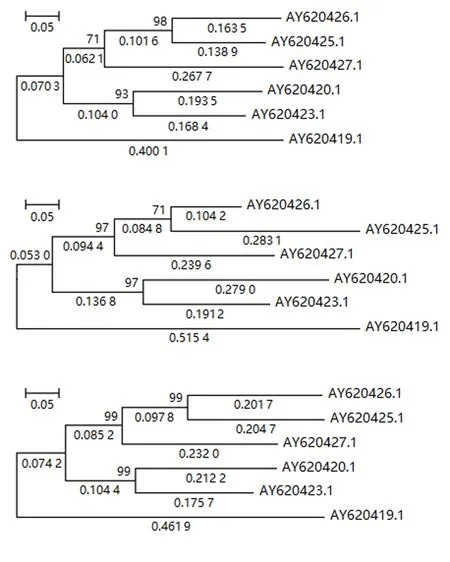
图4 第2组数据在3种预处理后重建的系统发育树Fig.4 Reconstructed phylogenetic trees from processed data of group 2 by 3 preprocessing methods
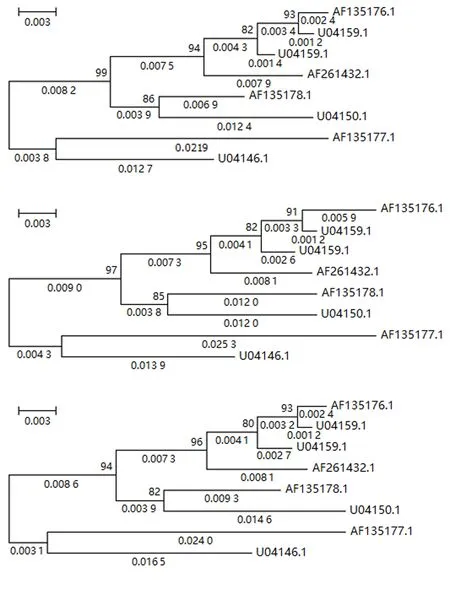
图5 第3组数据在3种预处理后重建的系统发育树Fig.5 Reconstructed phylogenetic trees from processed data of group 3 by 3 preprocessing methods
2.2.3 模拟系统发育的真实树与重建树枝长的误差 3种预处理下模拟同源序列系统发育重建树的枝长与模拟真实树枝长的误差统计结果(表4)表明:MNI和MLI插补的预处理方法下的枝长误差,无论是绝对误差还是均方根误差均小于Del预处理下的枝长误差,这是直接删除含有gap位点丢失进化信息所导致的.而MNI和MLI两者的枝长误差较小,且基本相同.经统计,在30组模拟序列的MNI和MLI之间存在7.556%±2.366% gap位点插补核苷酸的差异,这种差异在不同分枝枝长估算结果上体现出来.
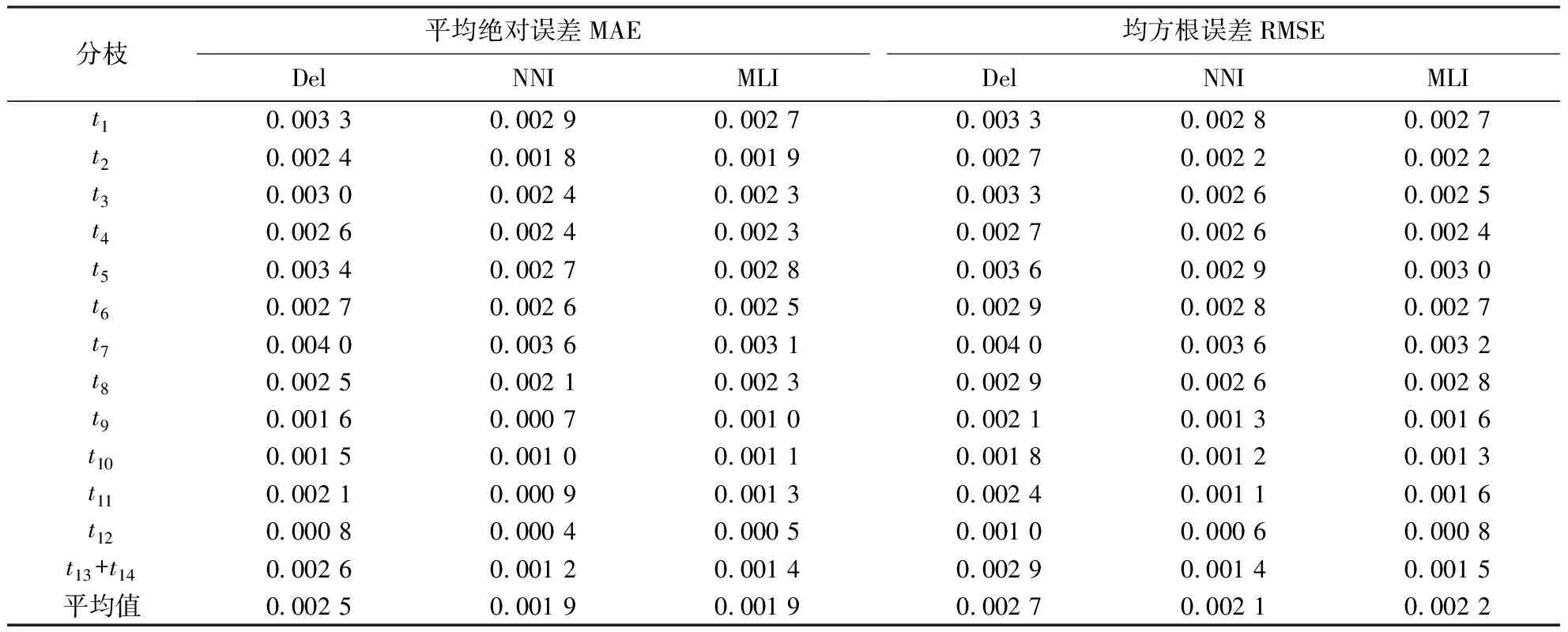
表4 重复模拟的系统发育树枝长在3种数据预处理下的误差Table 4 Deviation of estimated branch length of simulated phylogenetic trees under 3 preprocessing methods
3 小结
真实序列和模拟序列试验均证实了MNI和MLI两种核苷酸插补算法能够有效减小序列间进化距离的偏低估计;在系统发育重建中,两种核苷酸插补的预处理方法能够保持重建进化树的拓扑与流行的预处理方法一致的结构,并能提高枝长估算的精确度.而本文提出的核苷酸最大似然插补法与最近邻插补法在枝长估计误差的减小程度上基本相同,虽然最近邻插补的算法简捷,其时间复杂度[O(ms)]低于最大似然插补算法[O(4m-1s)],但鉴于最大似然法良好的统计学性质,推测其插补预处理后的数据更可靠.
