理论研究晶体场效应和电荷转移效应对Co2+的2p电子X射线L2,3吸收边光谱的影响
2021-07-11BORADebajeetGLANSPerAndersGUOJinghua
程 效,BORA Debajeet K.,GLANS Per-Anders,GUO Jinghua,罗 毅
(1.中国科学技术大学合肥微尺度物质科学国家研究中心,合肥230026;2.Advanced Light Source,Lawrence Berkeley National Laboratory,Berkeley CA 94720,USA)
1 Introduction
Cobalt-containing materials are receiving renewed interest due to their exotic properties originated from cobalt atom’s partially filled 3dshell in various potential applications,such as renewable energy[1—3],magnetism[4,5],spintronics[6],and catalytic reactions[7—14].Because of the exceptional element-selectivity and locality,X-ray absorption spectroscopy(XAS)is a powerful technology to probe electronic structure of the selected transition metal element in materials of interest.In particular,theL-edge XAS can in principle give the detailed information about the fundamental character of the 3dorbital and the bonding property between the centrally located transition metal and neighboring ligands.The-edges in various cobalt compounds have been extensively studied by different experimental methods,such as soft X-ray absorption spectrum[15—17],X-ray magnetic circular dichroism(XMCD)[18—20],and electron energy loss spectroscopy(EELS)[21—23].Since in various materials the ligands surrounding cobalt cation are either of different kinds or of different symmetrical local structures,the corresponding XAS profiles often show different features with unique characteristics.It becomes essential to find out connections between these specific spectral features and the local electronic structure property around cobalt cation.However,such rich spectral features appearing in the XAS are very difficult to interpret or assign by physical intuition.Advanced theoretical modeling is highly desirable.In this context,multiplet calculation method[24]has been quite successful to explain the interesting features found in the X-ray absorption spectra,especially some new features found in the novel materials[25].The multiplet calculation method uses crystal field cluster model combined with charge transfer multiple configuration interaction to compute the corresponding X-ray absorption spectra,which naturally connects the spectral feature with local electronic structure.
For cobalt containing materials,the multiplet calculation method is an effective tool to interpretedges X-ray absorption spectra[26—28].The multiplet calculations of Co2+L-edge XAS reveal many unique and interesting physical mechanisms,such as the important role of 3delectron spin-orbit coupling interaction[29,30],the possible mechanisms in chemical reactions[12—14],the origin of magnetism in cobalt doped materials[5,31].The sensitivity of cobalt(Ⅱ)carboxylates XAS to different ligand field has been studied experimentally and theoretically[32].The difference between the XAS of CoO nanocrystal and single crystal was analyzed by crystal field multiplet calculations and the effects of 3delectron spin-orbit coupling,tetragonal distortion,and superexchange interaction have been revealed[33].
The calculation method based on multiplet effects[24,34]used to study the-edges X-ray absorption spectra[26—28]is a semi-empirical method[35].It requires a good set of semi-empirical parameters for the calculation to describe the crystal field effect and charge transfer process.From as early as 1990s,certain set of multiplet calculation parameters have been found to be preferable for various Co2+systems in X-ray spectroscopy calculations[27,36].In the case of Co2p L2,3-edges XAS due to a number of low-energy excitation factors such as crystal field distortion,3delectron spin-orbit coupling,and temperature effect on population of the excited states,the physical meaning of the parameters and their specific effect on XAS have not been understood well in the previous researches.Therefor rather than to find the better calculation parameter set for certain system,in this work we provide a systematic study of validating the change of-edges X-ray absorption spectrum with respect to different calculation parameters variation.
The most common type of Co cation’s local structure found in the transition complex is Ohor close to with slight distortion such as CoO and CoCl2.In experimental findings specifically related with CoO and CoCl2,the-edges XAS were carried out to study self-assembled pencil-shaped CoO nanorods[37],electron correlation and charge transfer effect at Ni/Co interface[38],Co2+low spin state in individual Ti0.8Co0.2O2nanosheet[39],formation of CoO at Co2FeAl0.5Si0.5/MgO interface due to high annealing temperature[40]and the atomic structure of CoO nanoparticle as active catalyst for light-driven water oxidation[33].This research will focus on Co2+cation in the Ohsymmetry using CoO and CoCl2as example systems to study the variation ofX-ray absorption spectrum with respect to the change of different calculation parameters.The discussion made on Co2+cation in Ohsymmetry in this research is not only limited to CoO and CoCl2systems,but also can be applied to other similar materials with the same Ohsymmetry.
Although the Co2+cation share the same Ohsymmetrical local structure in CoO and CoCl2,there are differences between CoO and CoCl2structures.The different structure results in different crystal and charge transfer effects therefor requires different appropriate calculation parameters in multiplet method.As for CoO at room temperature,it is paramagnetic within acubic crystal structure,where Co2+cation sites in an Ohsymmetrical local structure surrounded by six O2-ligand anions.It becomes an antiferromagnetic material with a slight tetragonal distortion for Co2+below its Néel temperature 290 K[41,42].The CoCl2always exists in hydrate form with layer by layer structure,where in each layer Co2+cation sites in Ohsymmetrical local structure formed by six Cl-ligand anions and H2O intercalate between two layers.In other CoCl2hydrate forms or anhydrous form,the local structure around Co2+cation has slight distortion but can still be approximated by Ohsymmetry.
The configurational energy,Slater-Condon parameters and spin-orbit coupling interactions are calculated inabinitiomethod for 2p63d7as initial state and 2p53d8as final state.Then the crystal field and charge transfer effects are described by several semi-empirical calculation parameters in the following steps.When the crystal field is in the Ohsymmetry,the 3dorbitals of Co atom are split into two groups,t2gandeg.The parameter 10Dqis defined as energy splitting betweent2gandegorbitals and is used to describe the crystal field effect in Ohsymmetry.While the charge transfer effect is treated by inclusion of more configurations in the initial and final state,in a similar manner to configuration-interaction(CI)method.Charge transfer energy(Δ),on-siteCoulomb repulsion(Udd),core-hole potential(Upd),and hopping parameters(T)fort2gandegare used to describe the charge transfer process.The physical meaning of all these parameters used in the calculation will be discussed in details below.
The multiplet calculation on Co2+cation’s 2pX-ray absorption spectrum is performed in atomic model first,then the crystal field and charge transfer effects in Ohsymmetry are introduced into the calculation.The variation of XAS features with respect to the change of 3dspin-orbit coupling,crystal field strength,charge transfer energy,on-siteCoulomb repulsive interaction between 3delectrons,Coulomb interaction between 2pcore-hole and 3delectron,hopping parameters for different 3dorbitals and the temperature effects are discussed systematically in this work.The physics meaning of each parameter is revealed to elaborate the connection between the specific property of Co2+local structure and certain feature in XAS.The analysis routine in the work is not only limited for Co2+cation in Ohsymmetry,but also provides a systematic way on how to apply the multiplet calculation method of X-ray absorption spectrum of other transition metal systems.
The conventional multiplet method mentioned above is one of the most successful methods for computing X-ray spectra of metals.However,its quality is much dependent on the choice of several semi-empirical parameters we are going to discuss here.In recent years,there are several methods prompted for first-principle calculation of these multiplet theory semi-empirical parameters.Josefessonetal.[43]incorporated atomic multiplet and molecular orbital effectsviaconfiguration interaction for transition metalL-edge X-ray spectra in aqueous ions and molecule.The ligand field DFT method[44,45]implemented in the amsterdam density functional(ADF)program package was used to calculate the multiplet structure of some heavy metal compounds such as Slater-Condon integrals,spin-orbit coupling constants and ligand field potential.The DFT method[46,47]incorporated with Wannier functions is able to provide realistic values for parameters in Anderson impurity model,such as state energies,hopping parameters and Coulomb interactions in transition metals.Many other DFT methods[48—51]with Wannier functions were also reported to be able to determine non-empirically the multiplet energy levels and ligand-field parameters in rare earth elements.What’s more,latest plethora of reviews about different methods used to interpret 2p L-edge XAS of transition metals are presented by various experts in this field[52—54]both in semi-empirical and first-principle codes.Here in present study we just focus on the conventional crystal/ligand-multiplet approach’s semi-empirical parameters physical effects on spectral features,in the future we will introduce the first-principle calculation method to derive these parameters as many as possible non-semi-empirically.
2 Theory and Computation Details
The multiplet calculation method is based on Cowan-Butler-Thole code,based on a serial of development of many researchers’work since early 1960s[55—57],including the integration of CTM4XAS software package[35]by Frank de Groot’s group.In the calculation the crystal field effect is taken into consideration by using a symmetry branching method based on group theory.The charge transfer effect is incorporated by including more charge transfer configurations into the initial and final states.In the process of interaction of X-ray with materials,Fermi Golden rule plays an important role in absorption mechanism.The intensity of absorption spectrum(IXAS)of a transition from initial stateto final stateby absorbing an incident photon with energyωphis given under first order dipole transition approximation as[58]:
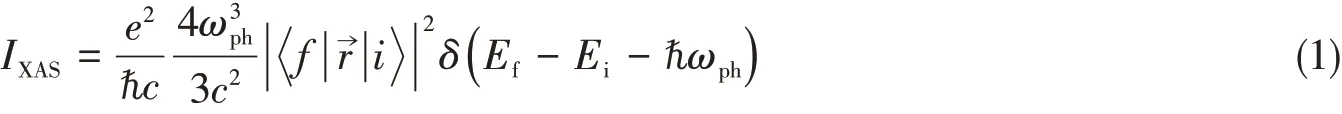
whereEiandEfare energies of initial state and final state,respectively;eis electron charge.
In order to calculate the wavefunctions of the initial and final states,the atomic multiplet calculation is taken under the single impurity Anderson model[59]using Hartree-Fock method following the method introduced by Cowan and modified by Thole[55].In this first atomic model approximation,the system Hamiltonian concerning the center-sited cobalt cation is written as:

wherepiis momentum ofi-th electron,Zis center metal atom number,Nis total number of the electrons,riis the vector ofi-th electron,rijis the distance betweeni-th electron andj-th electron,ξiis the spin-orbit coupling constant ofi-th electron whose orbital angular momentum isand spin angular momentum isBy solving this equation,the wavefunctions and the Hartree-Fock values of Slater-Condon-Shortley parametersFiandGi,average energy for each configuration and spin-orbit interaction constants are calculated as shown in Table 1.In the Hartree-Fock calculation the energy matrix is diagonalized and gives out energy levels for ground and excited states with atomic term symbol2S+1LJ.
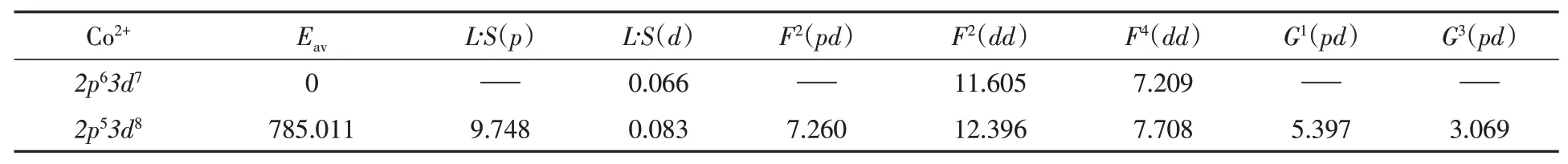
Table 1 Parameters from Co2+atomic multiplet calculation results*
For the following calculation steps,Slater integralsFiandGiare all scaled by a 80%factor compensating the excessive electron repulsive interaction calculated from the atomic model so as to give out a better simulated spectrum[27,60].When there is no crystal field,the pure isolated Co2+cation is treated in atomic spherical SO3symmetry group.The Ohsymmetrical crystal field is treated as a perturbation complement to the atomic Hamiltonian(Hatomic).The inclusion of crystal field effect with specific symmetry needs to branch the crystal field perturbation term from SO3group to Ohgroup following Butler’s method[61].The potential of crystal field effect(Hcf)can be written as a serial multipole expansion of spherical harmonic functions and symmetricalJmfactor parts as below[61,62],

whereMLis the number of the ligand anions around Co2+cation,Zmis them-th ligand anion’s charge,andR→mis the vector ofm-th ligand anion,is the spherical harmonic functionsis the conventional crystal field parameters,X kαβis the coefficient for operatorU kαβof the symmetrical branching chain SO3→G→g(G and g are symmetry symbols)from group theory[61].This equation indicates that the crystal field potential contributed from surrounding ligand anions(first equality)can be expressed in spherical harmonic functions form(second equality)which describes the spectrum terms.The further expression(third equality)is symmetrical branching method form which is used to describe the parameters needed for the code of Butler.By analysis of the equation above,one can tell that in Ohsymmetrical crystal field the five 3dorbitals split into two degenerated groups,threet2gand twoegorbitals.In the detailed calculation,the crystal field strength for this Ohsymmetry is represented as the energy splitting gap 10Dqwith a value between thet2gandegorbitals.
The charge transfer effect is then taken into consideration by introducing multiple configurations similar as configuration-interaction(CI)method.Normally,two configurations are appended in the initial state and final state.The Co2+ground state is then treated as 2p63d7+2p63d8L,whereLrepresents a hole in the ligand state.Then the configurational transition path becomes initial state→final state(2p53d8+2p53d9L).The Thole and Ogasawara’s code[63,64]is used to treat the charge transfer effect in the calculation.The parameters used to describe the charge transfer process are charge transfer energy(Δ),on-siteCoulomb repulsion(Udd)between 3d electrons,core-hole potential(Upd)between 2pcore-hole and 3delectron,hopping parametersT(eg),T(t2g)as hybridization energies between ligand 2porbitals and cobalteg,t2gorbitals.
The calculation parameters for crystal field and charge transfer effects are chosen literature values initially[27]and then change stepwise to show how the X-ray absorption spectral profile changes as function of each parameter.Finally the adjusted calculation is also compared with the experimental XAS spectra of CoO and CoCl2.In the calculation the Lorentzian broadening half width at half maximum(HWHM)value is set as 0.2 eV forL3edge and 0.4 eV forL2edge.The larger value forL2edge is due to Coster-Kronig decay of the 2p12core hole,a combination of life-time and phonon effect for difference Lorentzian broaden effect inL3andL2edges[65,66].The Gaussian broadening HWHM value for the whole spectrum is set as 0.2 eV.
3 Results and Discussion
3.1 Atomic Model Calculation
In the atomic multiplet calculation without crystal field and charge transfer effect,it is effective to describe the ground state of Co2+cation with the configuration2p63d7.According to Hund’s rules,the ground state atomic term for configuration 2p63d7is4F9/2with high spinS=3/2.
All dipole-allowed transitions from this ground state to theL-edge photon-excited states are calculated to give out the absorption spectrum as shown in Fig.1.Due to the strong 2pcore-hole spin-orbit interactionthe whole spectrum is divided into two regions,L2andL3edge regions.TheL3-edge region below 787 eV is attributed to transition 2p12→3d,and theL2edge region above 787 eV is attributed to transition 2p32→3d.There are 8 visible transitions from this ground state4F9/2to final states inL3region forming 5 peaks(R0-R4),and 3 visible transitions inL2region forming one peak(R5)and one ignorable transition at R5’s high energy side.The atomic multiplet calculation could provide a clear transition assignment for each peak,as shown in Table 2.It is obvious that the atomic multiplet calculation spectrum doesn’t fit with experimental data,which indicates it is essential to include at least crystal field effect in the calculation in order to be comparable with experimental spectrum.

Fig.1 Atomic multiplet calculation of Co2+2p L2,3-edges X-ray absorption spectrum
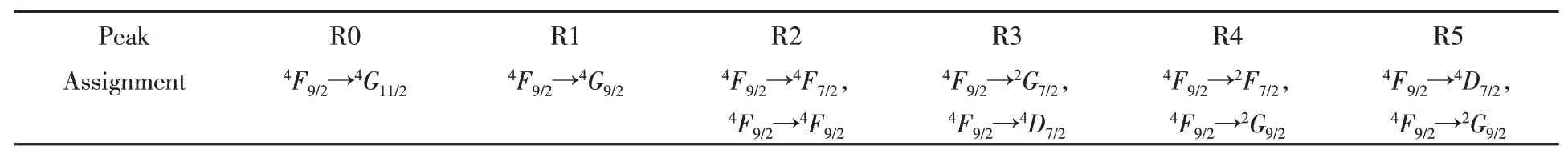
Table 2 Transition assignment of atomic multiplet calculation
3.2 Crystal Field Effect
When the Ohsymmetric crystal field is taken into consideration,the Ohsymmetry is processed by branching SO3→Ohaccording to the group theory[61,65].In this branching,the five degenerated3dorbitals of metal center in SO3group will split into two degenerated levels:eg(d z2,d x2-y2)andt2g(d xy,d xz,d yz)in Ohgroup as shown in Fig.2.The crystal field strength is represented by the energy gap value 10Dqbetweenegandt2g,which is the main control parameter for Ohsymmetric crystal field effect.The crystal field theory only takes the ligands around metal center as point charges.In order to incorporate the covalent interaction between ligands and central metal,the ligand field theory should be considered as an extension from crystal field theory.In the ligand field theory,the electron orbitals between Co2+and O2-are combination of Cobalt and Oxygen atomic orbitals.For the 3dorbitals of our study interest,the ligands O2porbitals are incorporated as group to combine with Co3dorbitals as shown in Fig.2.The O2porbitals from each ligand formsegandt2ggroup orbitals due to the Ohsymmetry as shown in Fig.2.Because of the same symmetry,the ligandegorbitals are able to overlap with Co3d egorbitals asσbonding to combine a bonding orbitaleg(σ)and an anti-bonding orbitaleg(σ*),while the ligandt2gorbitals are able to overlap with Co3d t2gorbitals asπbonding to combine a bonding orbitalt2g(π)and an anti-bonding orbitalt2g(π*).

Fig.2 Scheme of crystal field splitting and charge transfer effect
For different materials sharing the same Ohlocal symmetry for the center-sited metal cation such as CoO and CoCl2,the 3dorbitals splitting is the same but the different ligand anions around Co2+induce different crystal field splitting strength.The Tanabe-Sugano diagram(total energy diagram)of Co2+ground state configuration 2p63d7as shown in Fig.3,illustrate the multiplet states energy change at different crystal field splitting strength 10Dqvalues from 0 to 3.0 eV.The energies are calculated without 3dspin-orbit coupling for a simple view.The inclusion of 3dspin-orbit coupling interaction only makes current lines split into more states but the major shape of the lines remains the same.When 10Dq=0 eV,the Co2+is actually in atomic state where there are 8 states represented in atomic term symbol projection as shown in Fig.3.When Ohsymmetric crystal field is applied,these 8 states are split into about 20 states.As shown at 10Dq=3.0 eV in Fig.3,the 20 states are represented in Ohsymmetrical term symbol projection.In the process of 10Dqvariation,the multiplet states energy lines of labile electronic configuration 2p63d7have a singular change around 10Dq=2.77 eV,which is a typical spin-crossover phenomenon.The multiplet states change abruptly in the vicinity of 10Dq=2.77 eV,for example4T1→2Eand2E→4T1as indicated in Fig.3.
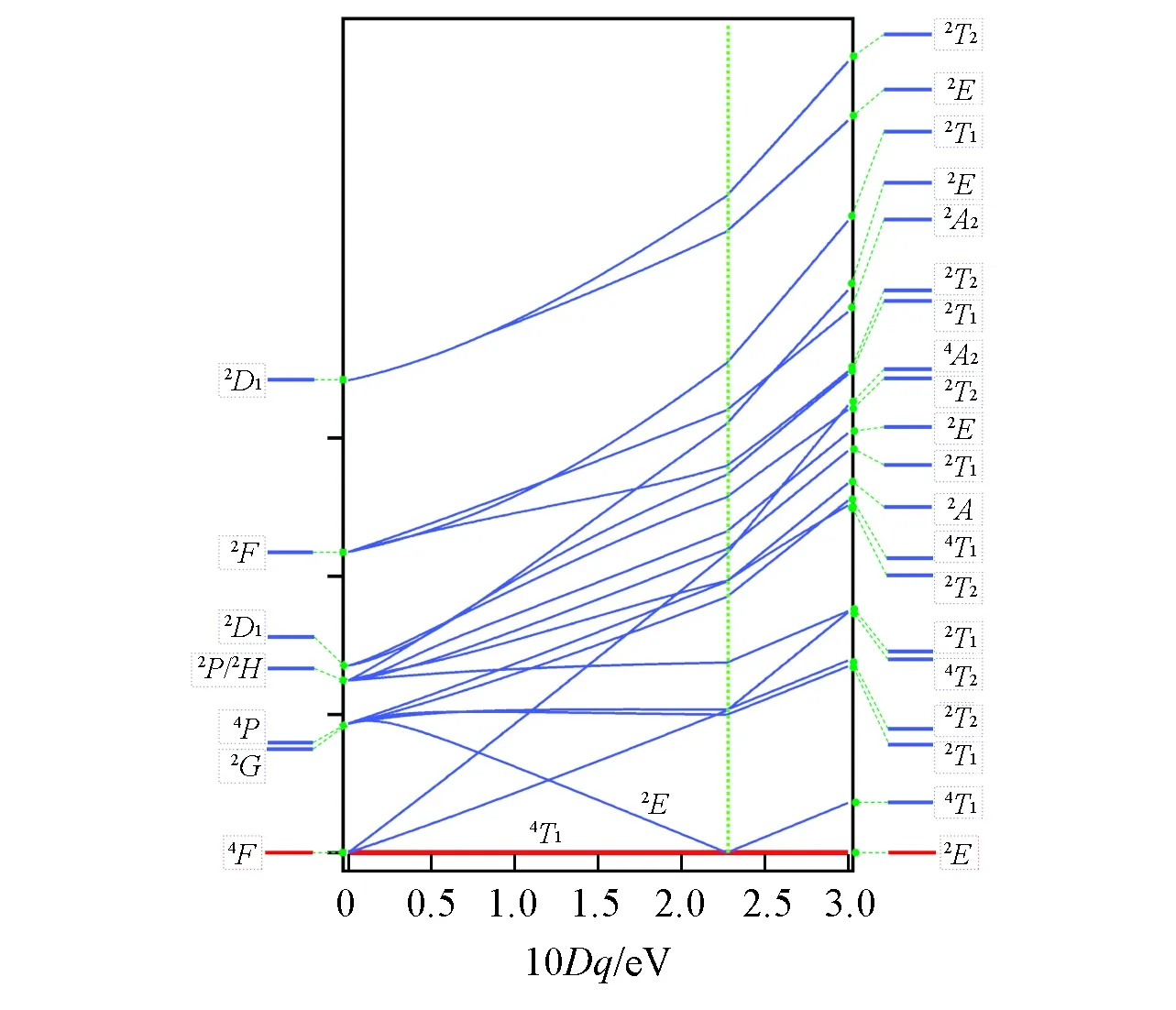
Fig.3 Tanabe-Sugano diagram of Co2+ground state without 3d spin-orbit coupling
As for CoO system,it is sufficient to choose 10Dq=1.0 eV for crystal field multiplet calculation without considering the charge transfer effect[29,36],the calculated spectrum could be close to the XAS observed in the experiment.The following discussion related to the effect of different physics parameters on XAS profile is not only for CoO and CoCl2,but can also be applied to all kinds of Co2+in an Ohsymmetrical structure with any possible ligands.
The variation of Co2+XAS spectra with 10Dqfrom 0 eV to 3.00 eV shown in Fig.4 demonstrates how different crystal field strength would affect the spectral features.The charge transfer effect is also included in this calculation and described by a set of parameters as used in the previous literature[27,36],where the charge transfer energyΔ=4.0 eV,on-siteCoulomb repulsionU dd=6.0 eV,core-hole potentialU pd=6.0 eV,hopping parametersT(eg)=2.2 eV,T(t2g)=1.1 eV.When the crystal field splitting strength 10Dqis weak,there are two degenerated unoccupiedegorbitals and one unoccupiedt2gorbital in ground state as shown in Fig.2.As the energy gap 10Dqbetweenegandt2gorbitals increases stepwise,the absorption energies for the photon excited 2pelectron transition to these unoccupied orbitals will also expand.As a result in Fig.4,there are more distinct multiplet structures inL2,3-edges as 10Dqincreases from 0 to 1.5 eV.

Fig.4 Electron distribution for high spin ground state 4T1 with small 10Dq(A)and low spin ground state 2E with large 10Dq(B)and variation of Co2+XAS spectra with 10Dq from 0 to 3.0 eV(C)
What’s more interesting is when 10Dqincreases over about 1.75 eV,a metamorphosis of the spectral profile is observed in Fig.4.This interesting phenomenon is because that large 10Dqcould induce a ground transition from the high spin(S=3/2)state4T1to the low spin(S=1/2)state2E.Typically the crystal field splitting strength 10Dqis relatively smaller than the inter-electronic repulsion energy,the electrons obey Hund’s first rule and the system prefers a high spin ground state.When 10Dqincreases over inter-electronic repulsion energy,the electrons tend to occupyt2gorbitals with lower energy and the system transfers into a low spin ground state,as shown in Fig.4(A)and(B).Thus,XAS can be a powerful tool for studying magnetic property of the material,for example to detect the artificial controlled spin states inter-conversion occurred in so-called spin crossover complexes[67].
3.3 Charge Transfer Energy
The charge transfer effect happens when one electron is transferred from ligand valence orbital to an empty Co3d t2gorbitalviaπbonding interaction as shown in Fig.2.The charge transfer energy(Δ)mentioned above is for the initial state,which defines the energy needed to excite an electron from ligand 2porbital to cobalt 3dorbital in the initial state.The charge transfer energy value depends on bonding property between ligand anions and center-sited metal cation.The higherΔimeans it is more difficult to have charge transferred from ligand to Co2+anion.For ground state with a configuration combination of 3d7+smallerΔiindicates more contribution from configurationwhile largerΔigives more contribution from configuration 3d7.SinceΔicontrols the weight of configurations in ground state,it can be considered as charge transfer initial state effect.
In Fig.5(A),XAS features vary with charge transfer energyΔifrom-7.0 eV to 7.0 eV,demonstrating how different electronegative ligands would affect the XAS spectral shape.WhenΔiis at a relative large value such as 7.0 eV,the charge transfer effect is very weak due to that the weight ofin the configuration combination is ignorable.IfΔicontinues to increase,the corresponding XAS profile will become more like pure 3d7calculation result in which only crystal field dominates.The charge transfer effect appears gradually in the XAS features,as the charge transfer energyΔidecreases from high value.The weight of 3d7andin the configuration combination becomes comparable to each other.The strong configuration interaction will result in dense mixture of the multiplet states of 3d7andin ground state[24].There will be two specific features appearing in the XAS profile due to this enhanced charge transfer effect.First,multiplet peaks afterL2,3-edges are observed in Fig.5(A).Second,multiplet structure inL3-edge compresses,but it is different compared to the multiplet structure contraction caused by decreased 10Dqin Fig.4.WhenΔidecreases further,the charge transfer effect will be restrained due to the dominant weight ofin the configuration mixture.TheL3-edge continues to compress into one sharp peak.The profile of the XAS will look much like pure 3d8calculation result with only crystal field effect,ifΔidecreases further.
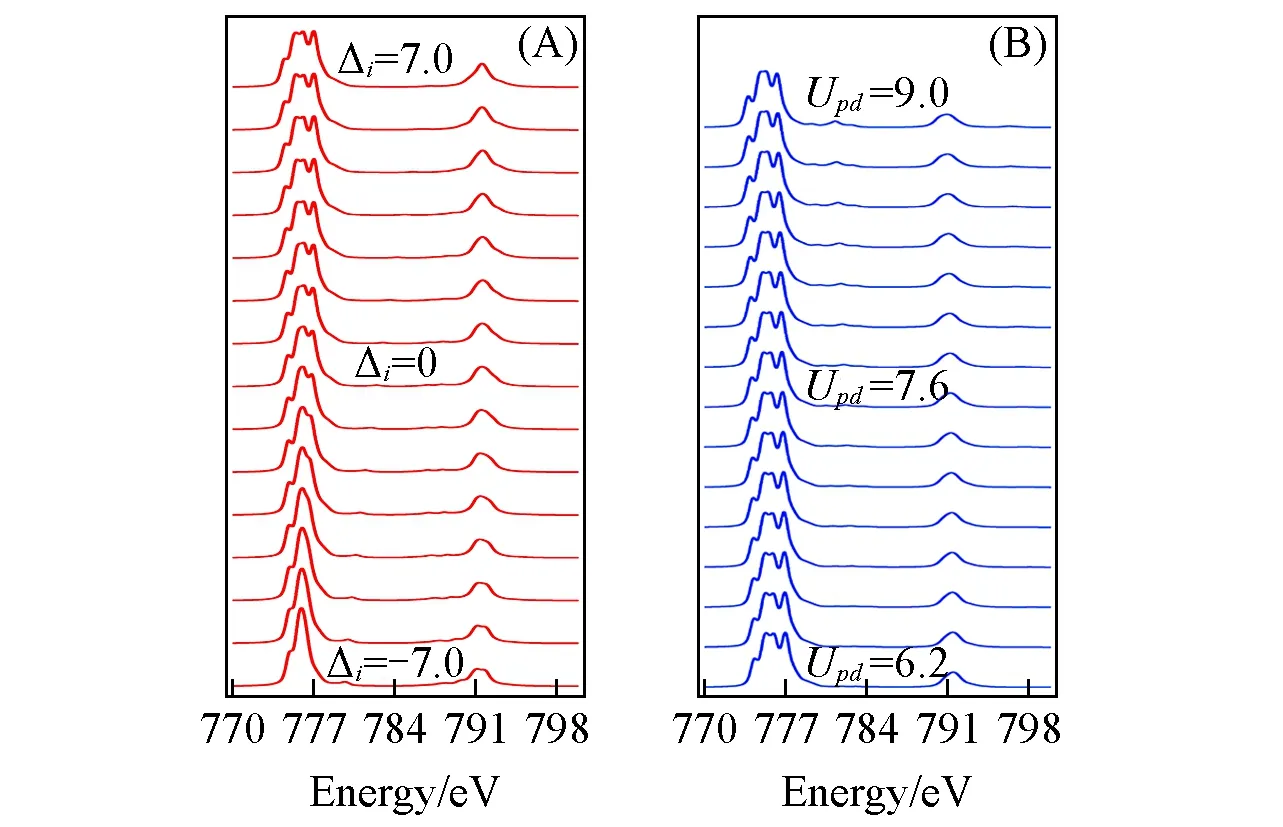
Fig.5 Variation of Co2+XAS with charge transferenergyΔi and core-hole potential Upd changes
3.4 The on-site Coulomb repulsion and Core-hole Potential
Theon-siteCoulomb repulsion(Udd)is also called HubbardUvalue,which is the lowest order term ofd-delectrons Coulomb repulsion integral.The core-hole potential(Upd)is the Coulomb interaction between 2pcore-hole and 3delectron.TheUddis also defined as energy difference[E(3d6)+E(3d8)-2E(3d7)],which describes the charge fluctuation from one Co2+site to the neighboring Co2+site in the impurity model.In the present approximation,the 2pcore-hole is only considered to have interaction with 3delectron.
Instead of considering each effect ofUddorUpdseparately,only the difference value ofUdd-Updwould affect the XAS spectral profile significantly,for exampleUdd=8.0,Upd=6.0 andUdd=7.0,Upd=5.0 give out the similar XAS spectral profile.The composite valueUdd-Updactually affects the configuration mixturein the final stateviaequationΔf=Δi+(Udd-Upd),when charge transfer energy of initial stateΔiis fixed.Compared toΔieffect mentioned above,the effect ofUdd-Updcan be considered as charge transfer final state effect.Similar asΔi,the small value ofUdd-Updmeans large weight of configurationin the final state.
In general calculation,theUpdis 1.0 eV or 2.0 eV larger than theUddparameter.WhenUpd-Uddvalue changes in a reasonable range(0.2,3.0 eV),the corresponding X-ray absorption spectra are shown in Fig.5(B)withUdd=6.0.The main shape ofL3-andL2-edges remains almost the same as theUpdchanges from 6.2 to 9.0 eV,but a satellite peak afterL3-edge around 781 eV and a weak satellite peak afterL2-edge appear gradually.The appearance of these interesting satellite peaks is due to the decreasedΔf=Δi+(Udd-Upd)inducing higher weight ofin the final state configurational combination,which gives a strong configuration interaction between them.The decrease ofUdd-Updindicates that theon-siteCoulomb repulsion(Udd)becomes weak while Coulomb attraction(Upd)between 2pcore-hole and 3delectron becomes strong.Both effects enhance the charge transfer from ligand or neighboring cobalt to center-sited Co2+cation’s 3dshell.As compared with the enhanced charge transfer processviaΔiinitial state effect as in Fig.5(A)where mainlyL3-edge profile changes,the increasedUpdhas a different final state effect on XAS profile as shown in Fig.5(B),where the weak satellites peak afterL2,3-edge appears with increasedUpd.The similar phenomenon was also observed in the XAS experiment of cobalt nanoparticles[25].In previous experiment and theoretical study[1],these satellite peaks were attributed toπback-bonding metal-to-ligand charge transfer(MLCT)effect,this work suggests that similar satellite peak may also be induced by enhanced charge transfer effectviaconfigurational interaction in final state.The previous effect is from the neighboring ligand coupling,but the latter one is due to the inner electrons interaction.
3.5 Hopping Parameters T(e g)and T(t2g)
The hopping parameterT(eg)orT(t2g)is defined as the hybridization energyV(L)between the 3dorbitalegort2gand the ligand valence orbital.In the charge transfer process,large hopping parameter yields high possibility of the electron transferred from ligand 2porbital to the certain 3dorbitalegort2g.When one electron transfers from ligand anion to center-sited Co2+cation,hopping parameterT(eg)orT(t2g)controls the weight of this electron transferred toegort2gorbital.Since the hopping parameter is always different for each group of degenerated orbitals,this would in some aspects contributes an additional crystal field splitting effect amongorbitals.
As shown in Fig.6,when the hopping parameterT(t2g)increases from bottom,the multiplet structure inL2,3-edges gradually compress.When one electron transfers from ligand state to Co2+t2gorbital,the resul-ted Coulomb block effect ont2gorbital leaves only two degenerated unoccupiedegorbitals available for the following photon-excited 2pelectron as described in Fig.2.The increase ofT(t2g)will enhance such process,and thus the absorption energies(Ef-Ei)become more degenerated,which reflects as compressed multiplet structure inL2,3-edges as in Fig.6.In contrast,when hopping parameterT(eg)increases from the middle,it broadens the multiplet structures in bothL3-andL2-regions as shown in Fig.6.When one electron transfers from ligand state to Co2+egorbital,the resulted Coulomb block effect onegorbital leaves only two non-degenerated unoccupiedegandt2gorbitals available for the following photon-excited 2pelectron as described in Fig.2.The increasedT(eg)will enhance such process,and results in more non-degenerated absorption energies(Ef-Ei),which reflects as broadening multiplet structure inL2,3-edges as in Fig.6.

Fig.6 Effeets of hopping parameters on XAS
3.6 Spin-orbit Coupling and Temperature Effect
As mentioned above,the 3delectron spin-orbit coupling plays an important role in Co2+L-edge XAS spectral profile.The calculated results for Co2+XAS with and without 3delectron spin-orbit coupling are compared in Fig.7.Unlike 2pcore-hole spin-orbit coupling which cleave the XAS profile intoL2-andL3-edges,the 3delectron spin-orbit coupling changes the features inL2,3-edges obviously.The 3delectron spin-orbit coupling interaction in Co2+will split the ground state4T1into four multiplet states with low energies.If the ground state withE2symmetry is set to be 0 meV,the first excited state with G symmetry has low excitation energy around 44 meV,the second excited state with G′symmetry is at 115 meV,and the third excited state withE1symmetry is at 128 meV[58].As a result,the temperature effect only applies on spectra with spin-orbit coupling interaction at high temperature over 200 K.The inclusion of low energy excited states by increased temperature makes the peaks inL2,3-edges merge together slightly in Fig.7.

Fig.7 Multiplet calculation of-edges with and without 3d electron spin-orbit coupling interaction
3.7 Comparison with Experimental Results
After the systematic discussion about each calculation parameter’s specific effect on XAS features,now we can apply the multiplet calculation to compare with the experimental results.Since Co2+ion in CoO and CoCl2share the same Ohsymmetrical local structure but with different local environments,they will be good systems to compare the correspondingX-ray absorption spectra.TheX-ray absorption spectra of CoO and CoCl2are collected in total electron yield mode as shown in Fig.8.The XAS profiles of CoO and CoCl2share the similar feature,with slight difference due to the same Ohlocal symmetry but different ligand anions O2-and Cl-.Different ligand anions O2-and Cl-have different crystal field strength and charge transfer effect in CoO and CoCl2.Compared to O2-anion,the Cl-anion has stronger charge transfer effect due to its stronger electronegative but weaker crystal field splitting strength.The calculation uses 10Dq=0.5 eV for CoO and 10Dq=0.4 eV for CoCl2to represent the stronger crystal field for O2-,and usesΔ=1.2 eV for CoCl2andΔ=2.4 eV for CoO to describe stronger charge transfer effect in CoCl2.All calculation parameters for CoO and CoCl2are listed in Table 3.Normally,U pdshould be 1 or 2 eV larger thanU dd,but in the present calculation for cobalt dihalides and cobalt oxide systems,it is appropriate to chooseU pdandU ddthe same value[27,28].The calculation results fit well with the experimental data of CoO and CoCl2respectively as shown in Fig.8.

Fig.8 X-ray absorption spectra of CoO and CoCl2 calculation results compared with the experimental ones

Table 3 Multiplet calculation parameters for CoO and CoCl2*
4 Conclusions
The parameters used in the multiplet calculation ofedges X-ray absorption spectrum in Ohsymmetry are discussed systematically,which revealed the physics mechanism behind each parameter and how they affect the XAS profile in specific pattern.The analysis method for each calculation parameter introduced here can be applied to other transition metals’multiplet calculation to get better XAS profile and understand new features appeared in the XAS of novel transition metal complexes.The multiplet calculation method is also applied on CoO and CoCl2as example systems with the spectra matching to the experimental results successfully.
