PD-1与单克隆抗体残基特异性结合机制的计算丙氨酸扫描研究
2021-07-11黄达锭鲍劲霄张增辉
温 炜,黄达锭,鲍劲霄,张增辉,2,3
(1.华东师范大学化学与分子工程学院,上海绿色化学与化工过程重点实验室,上海分子治疗与新药开发工程研究中心,上海200062;2.上海纽约大学计算化学联合研究中心,上海200062;3.美国纽约大学化学系,纽约10003)
程序性细胞死亡蛋白(PD-1)及其配体程序性死亡配体1(PD-L1)或程序性死亡配体2(PD-L2)是一种重要的免疫检查点[1,2],其在T细胞和肿瘤细胞中的表达上调可诱导免疫抑制[3,4].其中在肿瘤细胞中的表达上调就是癌细胞免疫逃逸的机制[5~7].基于阻断PD-1/PD-L1的单克隆抗体(mAb)免疫疗法显示出其能恢复病人的抗肿瘤免疫应答的作用[3,8,9].最近,2种靶向PD-1的单克隆抗体(Pembrolizumab和Nivolumab)已被美国食品和药物管理局(FDA)批准用于治疗黑色素瘤、非小细胞肺癌和颈部鳞状细胞癌[10].这些单抗在多发性肿瘤的临床应用中具有显著的肿瘤活性抑制作用[11~14].
虽然PD-1/Pembrolizumab和PD-1/Nivolumab的晶体结构已经分别由Lee等[15]和Tan等[16]确定,但是,关于有助于设计基于结构的单抗或小分子抑制剂的PD-1热点的详细信息仍然不清楚.因此,全面分析和了解这些单抗与PD-1的结合机制非常重要.再结合最近研究的PD-1与PD-L1在残基水平上的结合机制[17],将有助于下一代单抗的开发[18,19].
本文利用分子动力学研究了2个单抗(Pembrolizumab和Nivolumab)与PD-1结合的机制.使用计算丙氨酸扫描方法识别了PD-1/mAb复合物中的结合热点,对mAb和PD-1的结合热点进行了预测,并对两个体系热点的异同进行了分析和讨论.
1 实验部分
1.1 分子动力学模拟
从蛋白质数据库(PDB)[20]中下载了人源PD-1与抗体复合物(Pembrolizumab和Nivolumab)的2种晶体结构,其PDB编码分别为5GGS[15]和5WT9[16].用SWISS-MODEL服务器[21]对5GGS(PD-1上的残基25~30)和5WT9(PD-1上的残基85~93和重链上的残基128~133)中的缺失残基进行建模补全.所有动力学模拟均用AMBER14软件包[21]中的tleap模块对复合物进行氢原子的添加[22].Na+和Cl-被溶解在TIP3P水盒中以保持电中性,离子和蛋白的缓冲距离为1.2 nm[22,23].溶剂化后的系统用AMBER的ff14SB力场进行立场参数设置[23,24].为了消除边界效应,采用周期边界条件(PBC).采用Particle Mesh Ewald(PME)处理长距离静电相互作用,非键相互作用的截距为1 nm[25].步长时间设置为2 fs,用SHAKE约束包含氢原子的共价键[26].温度由Langevin恒温器控制,碰撞频率5.0 ps-1,压力由Berendsen气压表控制至1.01325×105Pa[27~30].
在能量最小化过程中,首先用2.09 kJ·mol-1·nm-2的力常数约束除氢原子以外的蛋白质复合物,然后用0.42 kJ·mol-1·nm-2的力常数约束复合物骨架,最后不用任何约束对整个体系进行优化.在加热步骤中,将每个系统从0 K加热到300 K,并以0.42 kJ·mol-1·nm-2的力常数限制在蛋白质主链原子上200 ps,300 K下在恒温恒容(NVT)和恒温恒压(NPT)系综中平衡22 ns.对蛋白质骨架重原子的均方根偏差(RMSD)进行了监测,使其在平衡阶段达到稳定状态.最后,进行了5次独立的22 ns动力学采样(轨迹),总模拟时间为110 ns,并选择最后2 ns轨迹进行能量分析.
1.2 MM/GBSA-IE-AS计算
对于每个复合物体系,丙氨酸扫描计算法[30~33]都用来计算残基特异性蛋白质-蛋白质结合自由能.本文扫描0.6 nm内的分子间残基对并将其突变为丙氨酸以计算PD-1/mAb复合物的野生型和突变型之间的结合自由能差在这项工作中,丙氨酸扫描(MM/GB/ASIE)方法[17,30~33]被用来计算单个残基对PD-1/mAb结合的自由能贡献.利用这种方法,相对结合自由能可以计算为

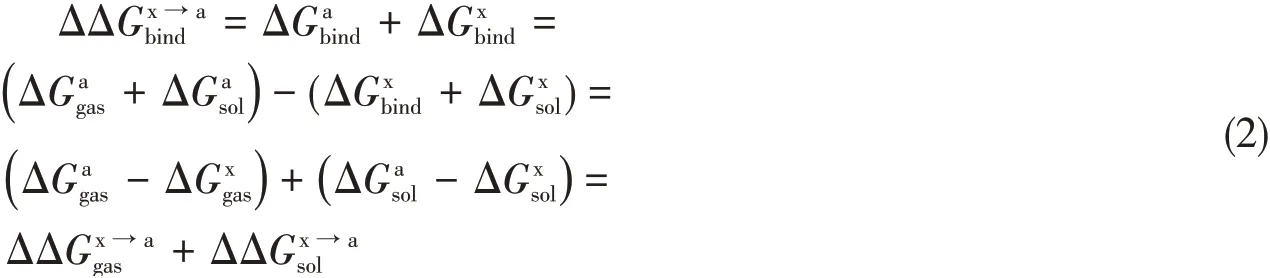
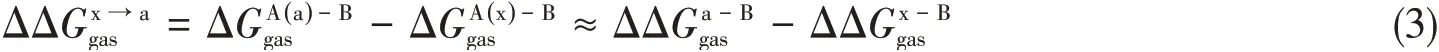
式中:A和B分别代表配体和受体;a和x分别代表突变后的丙氨酸和突变前的残基,残基的气相自由能贡献被近似处理成突变后残基和配体的气相结合能与野生型的残基与配体的气相结合能之差.在分子力学广义波恩表面积(MM/GBSA)方法[21]中,气相相互作用能定义为分子力相互作用焓和熵贡献之和,其中野生型的残基与配体之间的能量按下式计算[31]:

类似的,突变后的残基与配体之间的能量为
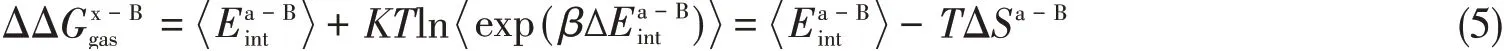

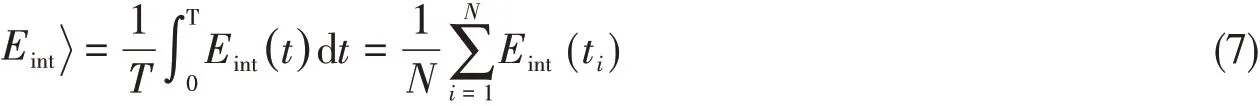
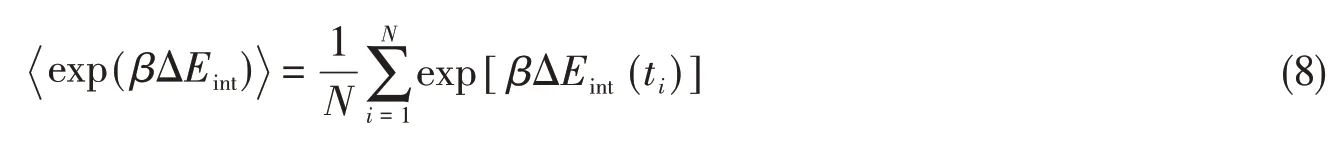

式中:γ和β值分别为2.26556 kJ·mol-1·nm-2和3.8456 kJ/mol的标准值[34].
对于每个PD-1/mAb复合物,运行5条MD轨迹,最终能量计算的结果是在5条独立轨迹上的取得的平均值ΔΔG(表1),这显示出单个残基对复合物结合的贡献.计算每个残基中的标准偏差(SD)以检验模拟误差(表1).在一条轨迹的GBSA/IE计算中,最后的2 ns轨道用于丙氨酸扫描计算.从轨迹中提取了10000帧,间隔为0.2 ps,用于MM/GB/ASIE计算.在AMBER14软件包中实现的MM/PB(GB)SA方法用于MM/GBSA计算[22].采用Yan等[32]使用的介电常数值,即非极性残基为1,极性残基为3,带电残基为5.在丙氨酸扫描法计算很多蛋白质-蛋白质的相互作用中,计算的范德华能量通常比实验数据高.因此,将芳香环残基的氢原子和碳原子的经验常数ε调整为AMBER14中PHE值的75%,TYP值的30%,TRY值的45%.MM/GB/ASIE计算的其它详细参数设置与文献[32]中相同.
Table 1 Computational alanine scanning result for the PD-1/pembrolizumab system
2 结果与讨论
2.1 结合复合物的动力学稳定性
通过原子位置的RMSD来测量PD-1/Pembrolizumab和PD-1/Nivolumab的5个系统的动态稳定性分别如图1(A)和(B)所示.彩色线表示骨架中所有重原子(非氢原子)相对于初始晶体结构的RMSD与模拟时间的函数关系.结果表明,复杂体系是稳定的,可用于计算丙氨酸扫描和其它分析.

Fig.1 RMSDs of the heavy atoms in backbone of the PD-1/pembrolizumab(A)and PD-1/nivolumab(B)complexes relative to the initial crystal structures
计算并绘制了PD-1/mAb体系中残基的均方根波动(RMSF),结果如图2所示.5WT9中PD-1缺失的残基85~93是PD-1/Nivolumab体系中最灵活的区域[图2(B1)],这些残基在晶体结构中没有确定坐标,是由SWISS-MODEL服务器模拟的.对于配合物中的PD-1,2种PD1/mAb体系中共有的最灵活的区域是残基71~74所在的loop区域[图2(A1)和(B1)].
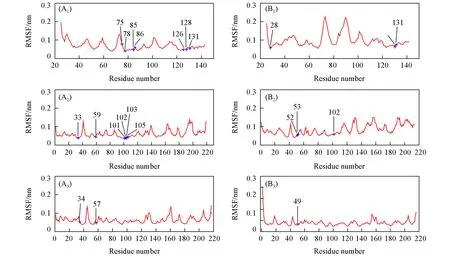
Fig.2 RMSFs of the Cαatoms calculated from the 22 ns trajectory of PD-1/pembrolizumab(A1—A3)and PD-1/nivolumab(B1—B3)trajectories
因此,用SWISS-MODEL服务器模拟的PD-1/mAb复合物结构(5GGS和5WT9)在MD模拟过程中是稳定的,轨迹用于进一步分析是可靠的.
2.2 PD-1上预测到的热点残基
MM/GB/ASIE法[31,32]用于计算2个体系(PD-1/Pembrolizumab和PD-1/Nivolumab)的残基特异结合能贡献,这2个体系在丙氨酸突变前后的离解速率常数实验值均不可用.在PD-1/Nivolumab复合物中,虽然Tan等[16]测量了4个N-连接糖基化位点(PD-1N49,PD-1N58,PD-1N74和PD-1N116)的实验数据,但PD-1在晶体结构中仅呈现出PD-1N58位点的糖基化信息,而在表面等离子共振(SPR)分析(用于测丙氨酸突变前后的离解速率常数)中包含4个糖基化位点,由于糖基化量的不同,SPR测到的丙氨酸突变前后的离解速率常数换算成的实验数据,不适合与只有1个糖基化位点的MM/GB/ASIE计算结果进行比较.图3(A)绘制了2个系统中PD-1的预测结果以用于对比.在2个系统中任意一个被预测为热点的PD-1上的残基被列在图3(A)中.共发现了9个此类残基,并将它们对结合配合物的相对能量贡献进行了比较.
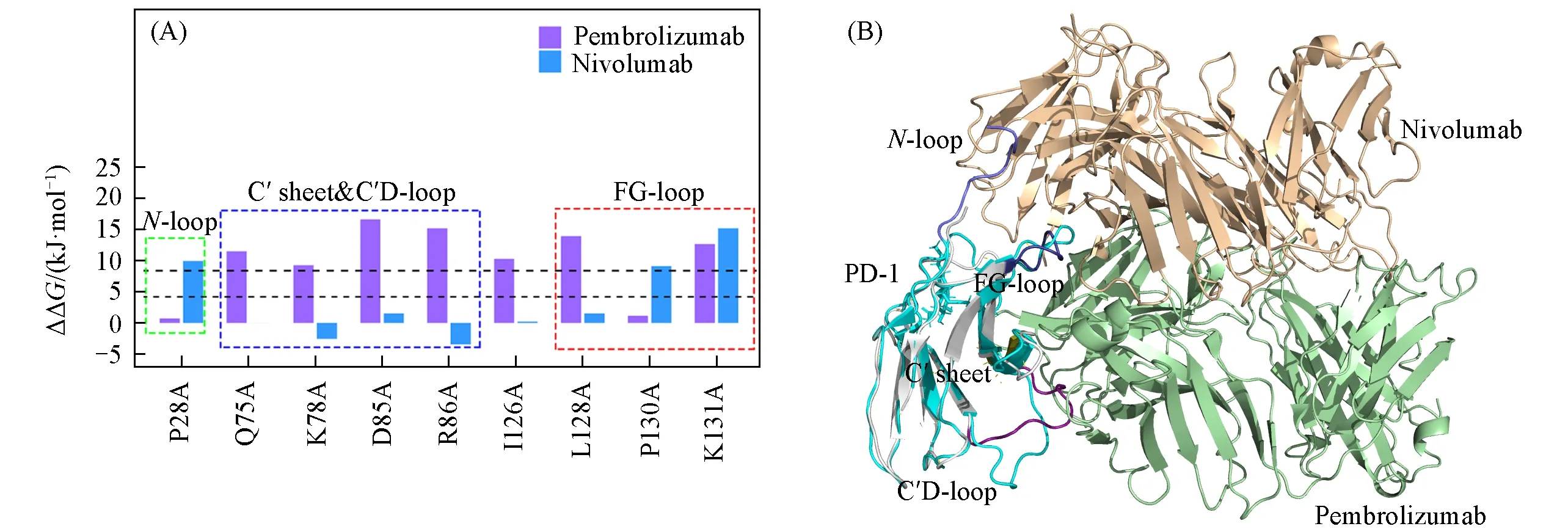
Fig.3 Hot spots predicted on PD-1 in two PD-1/mAb systems compared and arranged according to the sequence of residues on PD-1(A)and the superposition diagram of two PD-1/mAb complexes(PD-1/pembrolizumab and PD-1/nivolumab)(B)
2.3 2个PD-1/mAb体系中PD-1的特定分析
2.3.1 PD-1/Pembrolizumab 如图3(A)所示,PD-1D85是PD-1上最重要的热点.由表1数据可知,PD-1D85的自由能贡献主要来自静电能(ΔΔEele).PD-1D85和mAb-LR96之间形成强烈的静电相互作用[图4(A)].第2个和第3个重要的热点分别是PD-1R86和PD-1L128,范德华能量(ΔΔEvdW)占主导作用.

Fig.4 Details of the interaction of hotspots and warm spots at the PD-1/pembrolizumab(A)and PD-1/nivolumab(B)interfaces
2.3.2 PD-1/Nilvolumab 对于该体系,主要的相互作用来自带正电的PD-1K131,其与mAb-LY49形成了强烈的阳离子-π相互作用[图4(B)].此外,PD-1P28和PD-1P130也是重要的热点.如表2所示,vdW相互作用在所有3个热点中起着最重要的作用.
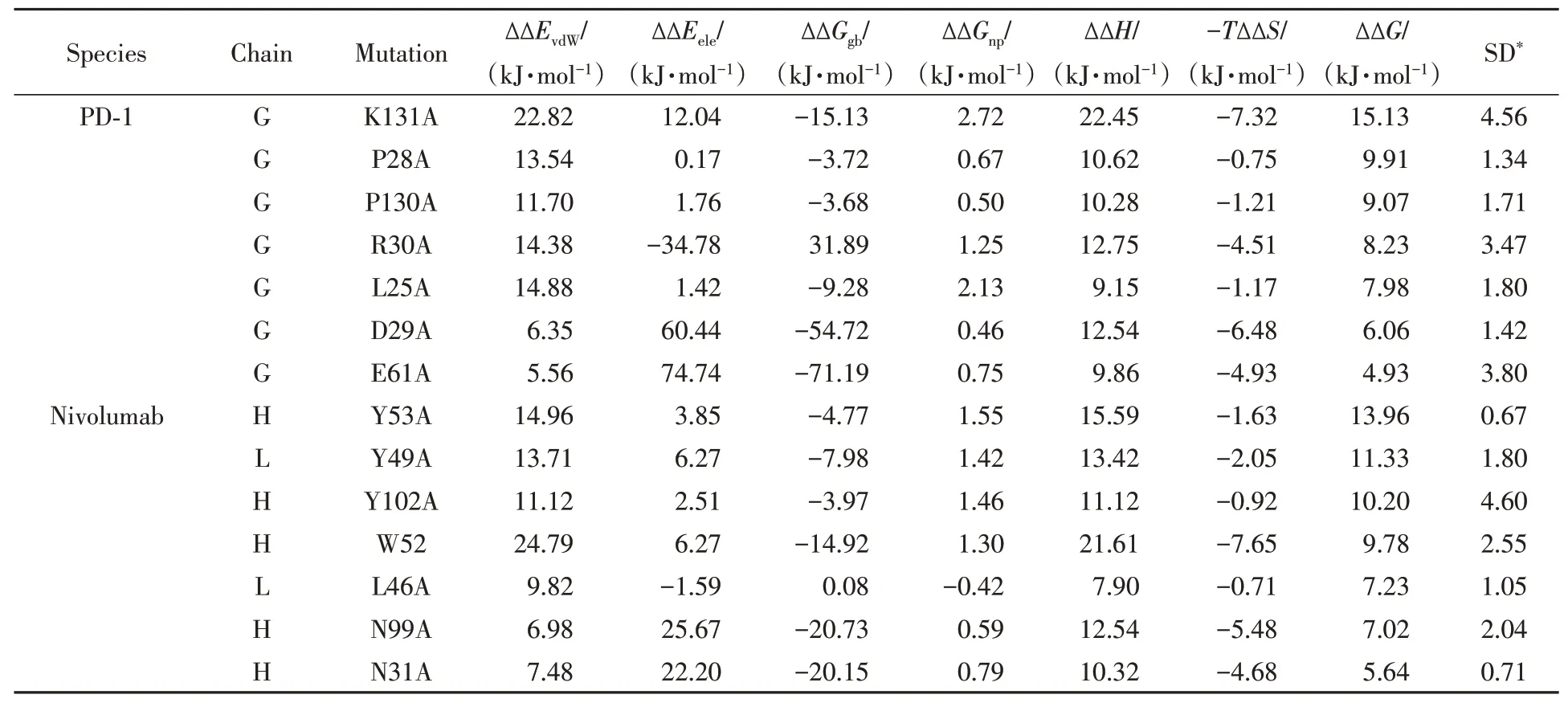
Table 2 Computational alanine scanning result for the PD-1/nivolumab system
综合来看这2个体系的最重要贡献是强疏水相互作用(图5),特别是阳离子-π、π-π或者methionine-π相互作用.唯一的例外是PD-1D85,其是PD-1/Pembrolizumab中最重要的热点,但PD-1D85的vdW能量为轻微的负值,静电能是主要贡献.这2个体系其它带电的热点残基都通过阳离子-π相互作用与抗体显示出强烈的vdW相互作用.从PD-1与单抗的结合区域来看,这两个系统中的差异较大[图3(B)].如图3(A)所示,2个系统中重叠的热点仅有PD-1K131.此外,如图3(A)和图4所示,Pembrolizumab主要与PD-1上的C′sheet(残基75~82)、C′D-loop(残基83~94)和重叠结合压(FG-loop)(残基127~134)结合[图3(B)],这与PD-L1和PD-1的结合模式[17]相似.然而,Nivolumab从不同的方向结合PD-1,结合区域包括PD-1上的FG-loop和N-loop[残基25~35,图3(B)][16].与此同时,N-loop在PD-1/Pembrolizumb的晶体结构中大部分是缺失的(晶体结构中该区域的缺失说明该处摆动特别大,基本没有结合位点).因此,从Pembrolizumab和Nivolumab与PD-1结合热点重叠少的结果可见,Pembrolizumab和Nivolumab之间存在不同的阻碍PD-1/PD-L1结合的模式,从FG-loop的角度来看,两者之间还可能存在一定的竞争关系[16].

Fig.5 Composition of free energy contribution for hotspots and warm spots predicted by MM/GB/ASIE method
2.4 单抗上预测的热点残基
因为在相互作用界面两侧进行了MM/GB/ASIE计算,所以除了分析PD-1的热点残基,还可以分析单抗中的关键残基.如图4和图6所示,对于PembrolizumabHF101是主要热点,其与PD-1K131和PD-1K78形成强烈的阳离子-π相互作用.HY33也是与PD-1K78形成阳离子-π相互作用的重要残基.在PD-1/Nivolumab复合物中,热点数量明显少于Pembrolizumab上的热点,这与PD-1/mAb系统中PD-1上的趋势一致.有趣的是,Nivolumab上的热点都是酪氨酸,Pembrolizumab上的热点酪氨酸所占比例最多,它们都是由vdW能量占主导的(图5).

Fig.6 Hot and warm spots predicted on the PD-1/pembrolizumab(A)and PD-1/nivolumab(B)
由于PD-1K131是前面对PD-1的分析中两个体系唯一共享的热点残基,因此研究了与PD-1K131相互作用的单抗残基之间的关系.如表3所示,与PD-1K131相互作用的残基几乎都有芳香侧链,它们的相互作用来自稳定的阳离子-π(PD-1K131的胺基离子与单抗的酚基).表3中与PD-1K131结合的残基包括4个酪氨酸残基,它们与PD-1K131的相互作用都相似地受vdW相互作用的支配.事实上,不只是与PD-1K131结合的残基,Pembrolizumab和Nivolumab上热点和温点的自由能贡献几乎均由vdW能量控制(图5).这说明提高抗体残基的静电相互作用的贡献,有助于进一步优化PD-1抗体的结合活性.
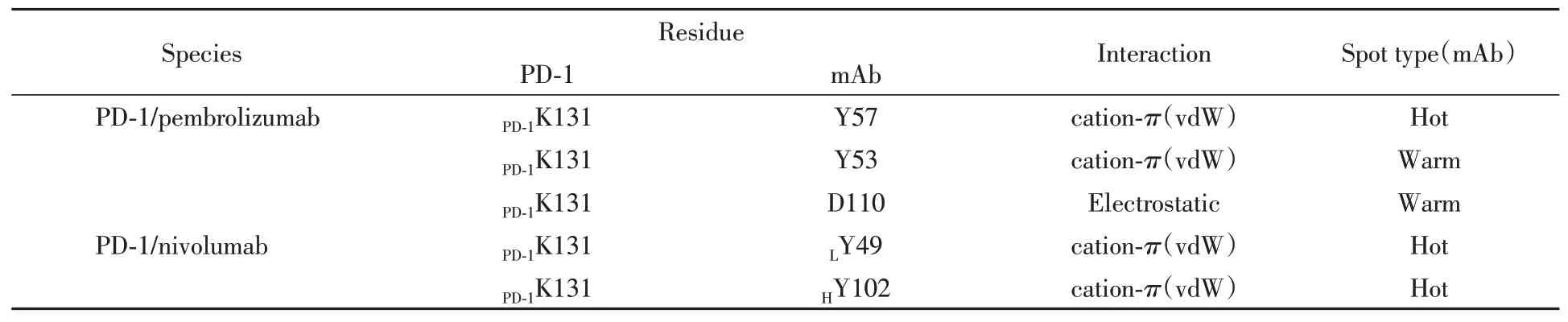
Table 3 Residues on the mAbs interacting with the only overlapping hotspots PD-1K131 and their main interaction types and the spot types of themselves
3 结 论
本文检测了2种PD-1单克隆抗体(mAb),并利用高效的计算丙氨酸扫描方法研究了它们与PD-1的结合机制.对PD-1/mAb结合起重要作用的热点残基进行了定量分析.对两种单抗与PD-1的结合方式进行了比较分析,同时对比了它们与PD-1/PD-L1结合方式的差别,发现PD-1和PD-L1与Pembrolizumab的结合方式类似,与Nivolumab的结合方式差异较大.PD-1K131是2个PD-1/mAb复合物中唯一有交集的相对重要的热点.在单抗方面,发现单抗与PD-1K131结合的关键残基以vdW相互作用为主,这也是整个抗体中最重要的相互作用贡献类型,本研究为设计以PD-1为靶点的新型单抗以及小分子药物提供了重要的启示.
感谢纽约上海和华东师范大学多功能创新平台(001)为我们提供计算时长.
