关于遗传基因检测中基因变异临床意义分级的建议
2021-07-09天津市医学会医学遗传学分会天津市医学会遗传咨询分会
天津市医学会医学遗传学分会,天津市医学会遗传咨询分会
由于近年来高通量测序技术,尤其是全外显子组测序技术的广泛应用,使得对海量的基因变异进行分析解读成为可能。美国医学遗传学与基因组学学 会(American College of Medical Genetics and Genomics,ACMG)发表了基因变异致病性的分类指南,包括非肿瘤性遗传病相关的胚系变异(为主体)的分类指南[1-3]、拷贝数变异的分类指南[4-7]、线粒体tRNA(mitochondrial tRNA)基因变异的分类建议[8-9]。近年关于致病性分类方法还在不断修订中[10-15]。在临床实践过程中,对于遗传基因检测报告的解读经常会产生误解,一方面源自变异水平的致病性分类方法尚不够完善;另一方面源自临床医生与实验室人员理解上的偏差。实验室专家往往强调基因变异的分类仅仅与变异本身的性质相关,而与具体案例(患者、携带者或者胎儿)的表型无关,而临床医生则需要直接面对临床案例,不仅要考虑变异本身的性质,而且要考虑案例的具体情况。
为了弥补实验室人员与临床医生之间关注焦点的差异,降低临床医生理解检测报告的难度,本文建议:基因检测实验室不仅要对变异的致病性进行分类,还应通过与临床科室的密切沟通,对疑似基因相关表型谱与具体临床案例之间的相关程度进行评估,并在此基础上对基因变异进行临床意义的分级。通过在检测报告中同时给出基因变异的致病性分类以及临床意义的分级,有助于临床医生准确地理解检测报告,也有助于下一步准确地向患者和(或)家属提供临床遗传咨询服务。
1 临床信息采集与疑似致病基因相关表型的回顾
1.1 详细表型和家族史采集的重要性 临床案例的详细表型和家族史对于后续基因检测结果的分析与解读具有极为重要的意义。表型资料与家族史的准确度和全面性不仅可能影响医生能否选择出最合理的检测项目,还可能影响到实验室在数据分析解读中能否定位到真正需要被关注的基因,也会影响到能否准确地进行致病性的评估。在致病性的评估过程中,一些证据点的评价需要考虑到临床相符的程度,如PP1(符合基因型-临床表型的共分离)和PP4(临床表型相符并且高度特异)[1]。因此,临床表型和家族史的采集是否完整与准确可能会影响最终的医疗实践。
在完成了基因检测之后,还可以根据基因检测报告所提示的疑似诊断再次进行表型采集,以便发现较为次要的表型,进一步确认或者排除诊断。尤其是针对携带可疑基因变异的家庭成员,也应尽量采集其表型,帮助对基因型-表型之间的相关性进行评估及确认。
1.2 辅助进行表型描述以及查询的数据库和网站
1.2.1 人类表型标准用语联盟(Human Phenotype Ontology,HPO)和中文人类表型标准用语联盟(CHPO)表型标准化数据库 随着对人类疾病研究的逐渐深入,科研工作者们越来越意识到临床表型数据的重要性,对基因型与表型数据进行联合分析成为众多疾病研究的一个方向。HPO旨在提供人类疾病中用于描述表型异常的标准词汇,目前包含约11 000个名词和超过115 000条关于遗传性疾病的注释,还提供了一套针对约4 000种疾病的注释(https://hpo.jax.org/app)[16-17]。HPO中每个术语描述了一种异常表型,如房间隔缺损。HPO建立者从医学文献、Orphanet、利用染色体组分资源建立的人类染色体不平衡与表型数据库(DatabasE of genomiCvarIation and Phenotype in Humans using Ensembl Resources,DECIPHER)和在线人类孟德尔遗传(Online Mendelian Inheritance in Man,OMIM)数据库获取信息并进一步开发,持续进行词条的维护和完善。HPO的建立有助于临床医生以标准化的医学术语来描述罕见病患者的表型,这样不仅有利于诊断疾病,确定致病基因,还能帮助研究人员寻找疾病与特定表型之间的关系。2016年初,国内相关领域的专家共同成立了CHPO,基于HPO网站进行词汇的中文翻译和编辑优化,并建立了CHPO搜索引擎(http://www.chinahpo.org),提供词库的免费下载,获得了国内同行的广泛关注和赞许。
1.2.2 OMIM数据库 OMIM数据库(http://www.omim.org)是关于人类基因和遗传紊乱的数据库,通过对新的病症分类并命名、收录表型和相关致病基因的关系来收录人类孟德尔疾病信息,可以通过临床特征、表型和基因型来搜索对应的信息。OMIM数据库包括大量已知的遗传病、遗传决定的性状及其基因,除了简略描述各种疾病的临床特征、诊断、鉴别诊断、治疗与预防外,还提供已知有关致病基因的连锁关系、染色体定位、组成结构和功能、动物模型等资料,并附有经缜密筛选的相关参考文献。OMIM数据库持续更新,是用来查询特定致病基因相关临床表型的较为可靠的数据库[18]。
1.2.3 GeneReviews数据库 GeneReviews数据库(https://www.ncbi.nlm.nih.gov/books/-NBK1116)是美国国立卫生研究院资助,由美国华盛顿大学组织编撰并维护的一系列在线丛书数据库。该丛书的编写目的是为繁忙的一线医学工作者提供可以直接在临床上应用的遗传病相关信息。GeneReviews内容非常详实,并支持遗传病外显率的查询。不仅如此,其还提供包括鉴别诊断、治疗方案、风险因素、监控方案和遗传咨询等信息,适合临床医生和遗传咨询师使用。截至2021年4月25日,GeneReviews共收录了796个章节且在不断增加中。2016年底,国内专家开始着手建立中文版GeneReviews(https://genereviews.nrdrs.org.cn/paper/index),翻译全部现有词条并根据英文原版及时进行更新,添加了更多英文版未涵盖的词条和中国人群特有的信息,如中国人群中的发病率和变异热点等。
1.2.4 人类基因突变数据库(Human Gene Mutation Database,HGMD) HGMD(http://www.hgmd.cf.ac.uk)存储了与人类疾病相关的致病变异信息,内容动态更新,不断添加新的数据,并且淘汰被认为可能存在错误的信息[19]。该数据库已经覆盖了超过1.1万个可能与人类疾病相关的基因,以及超过29万种有关的变异(以致病性的变异为主),并且注明可能与每个变异相关的临床表型。该数据库已经成为临床基因测序结果的解释中不可或缺的重要工具之一。
1.2.5 MalaCards数据库 MalaCards数据库(http://www.malacards.org)是人类疾病及其注释的综合汇编[20-21]。当前版本包括来自74个来源的21 369种疾病的信息。对于每一种疾病,数据库都会显示一张带有关于该疾病的各种注释信息的“疾病卡”,汇总了该疾病的各种已知信息。这些信息来自GeneCards数据库、文献检索和GeneAnalytics基因集分析工具。MalaCards数据库使用一个自动计算信息检索引擎,通过利用远程数据以及GeneCards平台收集的信息,整理和填充疾病卡。MalaCards数据库整合了专门疾病和一般疾病列表,包括罕见疾病、遗传病和复杂疾病等。MalaCards中每种疾病的相关基因的平均数多于OMIM,其特点是大而全,注解来源于文献,但可靠性不易判断。当在OMIM上未发现与基因对应的疾病时,可尝试该数据库。
1.2.6 DECIPHER数据库 DECIPHER数据库(https://decipher.sanger.ac.uk/index)是目前分子遗传学中最重要的生物信息学数据库之一。DECIPHER是一项国际化的合作项目,截至2021年4月25日,向该数据库上传数据的研究项目达到294个,开源的病例记录超过3.85万个。用户可以通过检索数据库发现一系列相关的遗传疾病信息,包括变异位点和临床表型等,从而提高临床诊断效能。
1.2.7 人类基因组结构变异(database of genomic variants,DGV)数据库 DGV数据库(http://dgv.tcag.ca/dgv/app/home)是正常人群的染色体结构变异数据库,提供了人类染色体结构变异的概况信息,记录了一系列基因变异与表型相关的信息,是目前在结构变异的致病性评级中常用的对照人群数据库。DGV数据库收录了健康人群样本中大于50 bp的基因组结构变化信息[22]。
1.2.8 MitoMap(人类线粒体基因组)数据库 MitoMap数据库(https://mitomap.org/MITOMAP)[23-24]收录了大量人类线粒体的全长序列数据(>51 800份)以及超过1.9万个基因变异位点,并对其中可能具有致病性的变异位点相关的临床表型进行了罗列。该数据库是临床线粒体病测序实验室最常用的数据库之一。
1.2.9 Phenolyzer工具 Phenolyzer工具(http://phenolyzer.wglab.org)可以十分方便地提供从标准化表型到可能相关基因的检索[25],目前已经被广泛应用于遗传分析工作中。该工具不仅能给出与特定表型(或一组表型)相关的基因信息,并且给出了按照优先级从高到低的排序。
2 基因变异致病性分类
2.1 基本的五分类 根据ACMG的相关指南,不论何种类型的遗传物质变异(点突变/拷贝数变异/线粒体变异),都可以按照五分类法对变异的致病性进行分类:致病性变异(pathogenic variants)、可疑致病性变异(likely pathogenic variants)、致病性不明确的变异(variants of unknown significance)、可能良性的变异(likely benign variants)、良 性 的 变 异(benign variants)[1]。
2.2 对致病性变异的细分 在解读致病性变异和可疑致病性变异时,还应当适时地考虑到外显率、表现度,甚至还需要考虑到致病机制。同一个致病基因的不同变异可能具有迥异的外显率和表现度,甚至还可以引起完全不同的表型。这些方面都是在基因检测结果的临床解释中要考虑到的。尤其是当患者家庭中存在变异携带状态符合患者的特点而没有临床表型的家庭成员时,对于外显率较低或者是极低的变异,应当尽量在临床报告中给出相关的信息。即使根据基因变异分类指南可以将此类变异分类为“致病性变异”,也推荐使用“外显率较低的致病性变异位点”、“外显率极低的致病性变异位点”或者“患病风险相关的基因变异”对此类变异进行描述。
对基因变异进行致病性分类时,还常常遇到一种窘境:由于临床信息可能不全面,或者不能确认患者与送检父母之间是否为亲生关系,使得本来可以获得较高级别的证据强度却不得不调低,仅能将变异分类为“致病性不明确的变异”,而在采集到完整的信息之后,恰好能根据更新的证据将致病性分类上升为“可疑致病性变异”。遇到此类情形时,推荐将此类变异的分类描述为“尚不能肯定具有致病性(variants of unknown significance,VUS),但偏向于具有致病性的变异(VUS-likely pathogenic,VUS-LP)”。
2.3 基因变异分类的扩展 针对可能与临床相关的疾病易感性变异位点而言,原则上应该综合是否已有功能学研究的证据支持、相关疾病(或特定表型)的遗传度、风险相关性的强度等因素。风险相关的强度可用比值比(OR值)、相对危险度(RR值)、加权遗传风险(CGR值)等指标来描述。本文建议,可根据是否已有功能学验证的证据将变异位点分为两类:一类位点不仅有群体统计数据支持,而且所在的基因与所关注的疾病之间的关系已经有充分的功能研究证据;另一类位点则仅仅有群体统计数据支持,但是尚缺乏可靠的功能研究证据支持。
尚有一类基因变异,它们可能有较高的人群频率,通常并不引起疾病,但是有可能会引起特殊的生化表型或者特殊的体征。此类变异可以划分为功能多态性变异的范畴。在特定的情况下,检测到此类变异也可能辅助临床解释。因此,在基因变异的分类中,除了按是否具有致病性进行划分以外,补充功能多态性变异也有助于临床实践和遗传咨询。见表1。

Tab.1 Pathogenicity classification of human gene variants表1 基因变异的致病性的分类
3 意外发现
在进行高通量测序,尤其是全外显子组测序和全基因组测序时,检测的范围不仅覆盖了与重点关注的表型相关的基因,也覆盖了其他大量的遗传信息。在对高通量测序结果分析时,常常可以意外地发现与重点关注表型无关,但是具有重要的提前诊断意义的基因变异。
ACMG最早在2012年开始了关于意外发现的讨论[27],并且后续提出了较为体系化的基本原则以及具体的“最少应该报告的意外发现的致病基因范围”[28-30]。作为基本原则,在高通量测序数据的分析和报告中,至少应该报告外显率较高、可引起严重损害、提前诊断可以使得患者获益的致病性的变异[29]。
4 案例的临床表型与可疑基因相关表型谱之间的相符程度
不论是基因变异的致病性分类,还是对案例的检测结果进行解释,实际上都需要对可疑基因相关表型与具体案例的临床信息(包括患者及家属的临床表型、家族史、变异的传递方式等)之间的相符程度进行评价。本文建议,将临床符合度划分为6个级别。
1级,高度特异性相符。第一类:临床案例具有某些高度特异的表型与疑似基因相关,仅1~2个基因与该表型相关,如有酶活性检测结果支持的、凝血因子Ⅸ/Ⅹ的活性减低的、具有高度特异性的代谢组学检测结果支持的。第二类:患者具有一系列特殊表型的组合,特殊的发育里程碑(某些遗传病的患者在不同的发育阶段表现出不同的特殊表型),仅1~2个基因与该表型相关,如符合Prader Willi综合征的里程碑及系列特征性表型、符合天使综合征的里程碑及特征性表型。
2级,具有一定特异性的相符。有部分特征表型,但是有3~5个相关基因,如眼皮肤白化病表型;或是有多个基因,但是其中某个基因占比超过50%的,如常染色体隐性遗传性非综合征型耳聋患者检测出1个GJB2的可疑变异。
3级,临床相符但是没有特异性。此类表型具有高度的遗传异质性,缺乏特异性,如智力障碍、自闭症、癫痫发作。
4级,临床部分相符,同时存在冲突但不能排除相关性。临床表型与疑似基因的相关表型有矛盾之处,但是可能用不完全外显、不完全显性、伴随其他疾病来解释。
5级,有冲突可排除。有部分表型相似,但是有某些表型完全不符,并且存在根本性的矛盾,不能解释。如临床疑似肌营养不良,但是免疫组化结果完全排除了抗肌萎缩蛋白病(DMD)的可能性;临床疑似糖原累积症,但是酶学检测酸性麦芽糖苷酶活性完全正常,可排除GAA致病基因的相关性。
6级,不相符。临床表型与疑似病因完全没有关系。
5 临床意义分级的办法
综合基因变异的致病性分类、临床相符程度之后,可进行临床意义的分级。
1级,很可能解释临床信息,或具有极强临床指导意义的变异。(1)对于案例的诊断有较为明确指导意义的变异。①显性遗传基因,分类为致病性或可疑致病性的变异,杂合子、半合子或纯合子(需确认半合子、纯合状态同样可以致病),外显率不低,临床相符度为1~4级。须注意,对于既可能显性遗传,又可能隐性遗传的基因,必须在变异水平确认遗传方式;纯合子患者的表型可能与杂合子个体的表型完全不同;个别显性遗传基因的半合子或者纯合子状态可能是不致病的。②隐性遗传基因,分类为致病性或可疑致病性的变异,复合杂合子、纯合子或半合子,外显率不低,临床相符度1~4级。③线粒体环DNA,分类为致病性或可疑致病性的变异,临床相符度1~4级。(2)可以完全解释特定临床表型的功能多态性位点。(3)对于患者的治疗或预后具有明确指导意义的变异。
2级,可能有助于解释临床,或具有潜在临床指导意义的变异。(1)对于案例的诊断有潜在指导意义的变异。包括:①显性遗传基因,分类为VUS,临床相符度为1级;②显性遗传基因,分类为VUS-LP,临床相符度为1~2级;③显性遗传基因,分类为VUSLP,临床相符度为3~4级(可能在获取了充分的表型信息之后将临床相符度上升为1~2级,将致病性分类上升为可疑致病变异);④显性遗传基因,分类为VUS-LP,临床相符度为1级,遗传自否认表型的父母中某一方,表明该变异存在不完全外显;⑤隐性遗传基因,纯合子,分类为VUS-LP,临床相符度为1~2级;⑥隐性遗传基因,复合杂合子,其中1个分类为致病性或可疑致病性,另一个分类为VUS,临床相符度1~3级;⑦隐性遗传基因,推测为复合杂合子但未经证实,其中1个分类为致病性或可疑致病性,另一个分类为VUS,临床相符度1~3级;⑧遗传方式相符,致病性分类为致病性或可疑致病性,但外显率较低,临床相符度1~4级;⑨遗传方式相符,致病性分类为致病性或可疑致病性,但外显率极低,临床相符度1~3级;⑩患病风险相关的变异,已有明确的功能研究的证据,临床相符度1~3级;⑪疑似的功能多态性变异,临床高度相关;⑫线粒体环DNA,分类为致病性或可疑致病性的变异,外显率较低,临床相符度3~4级。(2)可能解释特定临床表型的功能多态性位点。(3)对于案例的治疗或预后有潜在指导意义的变异。
3级,临床指导意义不肯定,且不能排除临床相关性的变异。(1)显性遗传基因的VUS-LP,临床高度相符,遗传自否认表型的一方父母,没有证据表明该基因存在不完全外显,或者没有证据表明外显率为100%。(2)隐性遗传基因,复合杂合子,分类均为VUS,临床相符度1~4级。(3)隐性遗传基因,纯合子,分类为VUS,临床相符度1~4级。(4)隐性遗传基因,推测为复合杂合子但未经证实,分类为VUS,临床相符度1~4级。(5)隐性遗传基因的一个杂合致病性/可疑致病性变异,临床相符。(6)致病性不明确、临床相关性亦不明确的拷贝数变异(Copy number variation,CNV)。
4级,意外发现的具有潜在临床干预指导意义的变异。(1)符合ACMG意外发现指南的变异。(2)本实验室自行定义的具有潜在临床干预指导意义的变异。
5级,与临床信息无关的变异,或者分类为良性或可能良性的基因变异。
6 临床意义分级的应用
本文所提出的临床意义分级,首先需要厘清基因变异水平的致病性评估与病例的临床意义的评估(图1)。本文建议,在基因检测报告中,不仅描述基因变异的致病性分类,而且应描述基因变异的临床意义分级,从而提高检测报告的可理解性。以下通过3个实际案例来阐述如何在临床检测中对基因变异进行临床意义的分级。
(1)案例1。女,4岁。因运动后出现行走姿式异常入院。母亲患有“多巴反应性肌张力障碍”,目前服用小剂量左旋多巴,可有效控制肌张力障碍发作。采集患者外周血进行全外显子组测序,检测到1个杂合变异:GCH1(NM_000161.2)Exon3:c.468T>A p.Tyr156*;后续检测患儿母亲的外周血,同样检测到该变异。
依据ACMG规范进行致病性证据描述与分类如下:该变异为无义变异(编码蛋白质的第156位氨基酸Tyr变为终止密码子,预计可使蛋白质翻译提前终止);HGMD、ESP6500、千人基因组和dbSNP147数据库均未见收录;患儿及母亲的临床表型均与GCH1基因相关表型相符。综合考虑,该变异为致病性变异(按照ACMG的相关证据缩写为:PVS1、PM2_supporting、PP1)。
依据本建议进行案例水平临床分级如下:GCH1基因的杂合性失功能性变异已经被反复报道可以引起多巴反应性肌张力障碍伴或不伴高苯丙氨酸血症(MIM128230),这可以解释患儿及其母亲的临床表现。综合考虑,临床分组为“1级,很可能解释临床情况,或具有极强临床指导意义的变异”。
(2)案例2。男,32岁。主因言语欠清1 d入院。近3个月脑出血2次,发现高血压2年,痛风5年,目前用4种降压药血压仍控制不佳。采集患者外周血进行全外显子组测序,检测到2个杂合变异:ABCG2(NM_004827.2)Exon5:c.421C>A;p.Gln141Lys;KCNH2(NM_000238.3)Exon12:c.2892delC;p.Gly965Glufs*9。
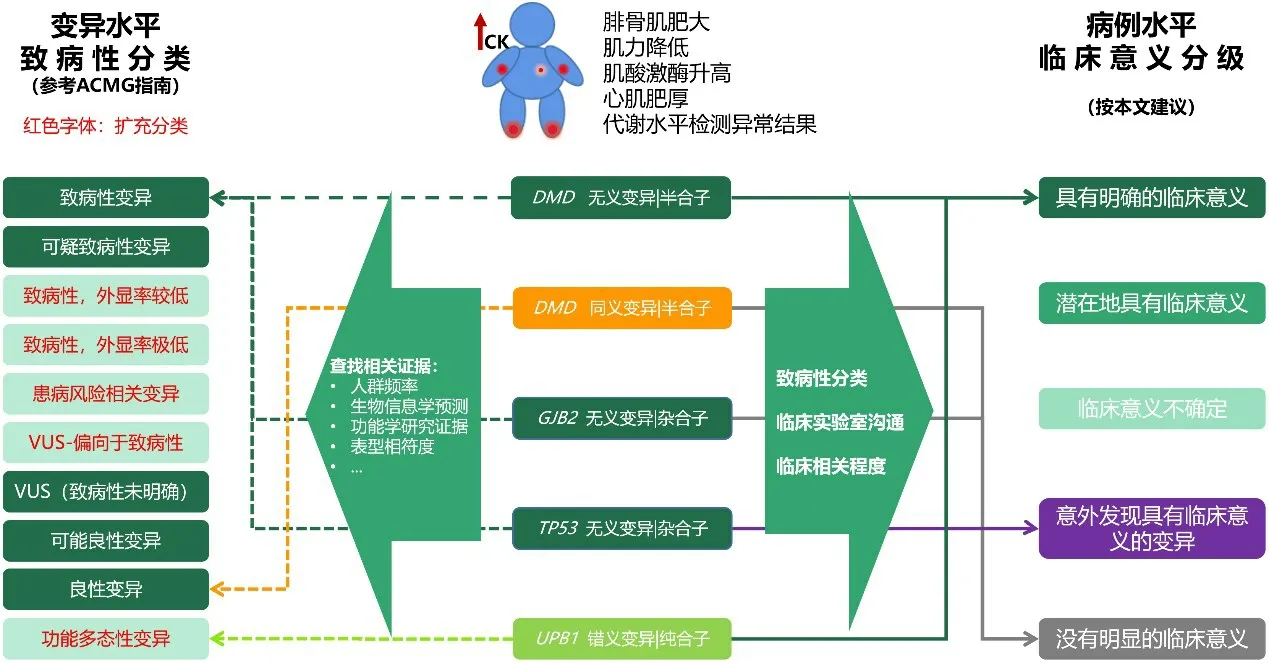
Fig.1 Variant level pathogenicity classification and case level clinical significance grading图1 基因变异水平的致病性分类与病例水平的临床意义分级
依据ACMG规范进行致病性证据描述与分类如下:(1)ABCG2基因变异(Gln141Lys)。大量文献支持该变异与痛风的风险相关,杂合子变异的个体患痛风风险提高1.74倍[31-33]。综合考虑,该变异分类为患病风险相关的变异。(2)KCNH2基因变异(Gly965Glufs*9)。该变异为框移变异,可引起NMD效应(无义介导的mRNA降解),导致基因功能异常,类似的变异被大量报道可引起长QT综合征;G1000、ESP、ExAC_ALL与gnomAD_genome_ALL数据库均未见收录。综合考虑,分类为致病性变异(按照ACMG的相关证据缩写为:PVS1、PM2_supporting、PP4)。
依据本建议进行案例水平临床分级如下:(1)ABCG2基因变异(Gln141Lys)。该变异已经被明确可以提高痛风症的风险。患者痛风5年,其他临床表现可能由痛风继发(临床相符但是没有特异性)。综合考虑,临床分组为“2级,可能有助于解释临床,或具有潜在临床指导意义的变异”。(2)KCNH2基因变异(Gly965Glufs*9)。该变异有害性明确,携带该变异患长QT综合征的风险较高,而尽早定期进行心电图等电生理检查及采取预防手段,极可能会减少或推迟发病,从而防止产生严重后果。综合考虑,临床分组为“4级,意外发现的具有潜在临床干预指导意义的变异”。
(3)案例3。女,2岁,弃婴。反复癫痫发作。采集外周血进行全外显子组测序,检测到SCN3A基因2个杂合变异:SCN3A(NM_006922.3)Exon8:c.905A>G p.Asn302Ser和Exon9 c.968G>T p.Ser323Ile。由于父母样本不可获得,故不确认2个变异的相位(incis或者in-trans状态)。
依据ACMG规范进行致病性证据描述与分类如下:(1)SCN3A基因杂合变异(Asn302Ser)。该变异为错义变异,有文献报道该变异可影响电压门控钠通道的功能[34];dbSNP147、ESP6500siv2_ALL和千人基因组(1000g2015aug_ALL)数据库均未见收录;生物信息学软件预测其有致病可能性。综合考虑,该变异分类为可疑致病性(按照ACMG的相关证据缩写为:PS3_moderate、PP4、PM2_supporting、PP3)。(2)SCN3A基因杂合变异(Ser323Ile)。该变异极为罕见,HGMD、dbSNP147、ESP6500和千人基因组数据库均未见收录;生物信息学软件预测其可能影响mRNA的剪接(dbscSNV_ADA预测值是0.962,dbscSNV_RF预测值是0.692),且预测其有致病可能性。综合考虑,尚不能确定该变异是否有致病性(按照ACMG的相关证据缩写为:PM2_supporting、PP3)。
依据本建议进行案例水平临床分级如下:(1)SCN3A基因杂合变异(Asn302Ser)。SCN3A基因与家族局灶性癫痫伴可变病灶4型(MIM617935)及婴儿早期癫痫性脑病62型(MIM617938)相关,均为常染色体显性遗传,相关表型有发育不良、喂养困难、局灶性癫痫、肌张力降低等,与该临床信息比较来看,临床相符但是没有特异性,该基因变异显性遗传且外显率不低。综合考虑,临床分组为“1级,很可能解释临床情况,或具有极强临床指导意义的变异”。(2)SCN3A基因杂合变异(Ser323Ile)。与前一个变异相比,其是否具有致病性尚不能确定,不排除该变异以in-cis或者以in-trans的状态与前一个变异存在协同作用的可能性。综合考虑,临床分组为“2级,可能有助于解释临床,或具有潜在临床指导意义的变异”。
7 其他
目前,绝大多数遗传病尚没有或者很少有特异性的治疗手段,但对遗传性肿瘤已经有一些基因变异可以指导此类患者用药。关于这部分变异的分类,也已在相应的ACMG指南中讨论。考虑到这类情况的存在,本文建议在遗传病的基因变异分类中,也采用相同的原则对具有治疗指导意义的变异进行分级,这对于遗传性肿瘤患儿而言是有价值的。由于对非肿瘤遗传病有治疗指导意义的位点为数不多,本文尚未对这些变异进行展开论述。在临床基因检测中如果遇到可指导治疗的基因变异,建议参照肿瘤变异分级指南[35-36]。
目前较为主流的临床全外显子组测序技术并未特别针对具有治疗指导意义的变异进行方法优化,这可能使得部分具有明确用药指导意义的变异位点不在覆盖范围内。全基因组测序技术已经在临床遗传基因测序实验室中有少量的应用[37],主要是针对比较严重的疑似遗传病患者进行检测,如多发畸形[38]和心脏疾病[39]等多种遗传病。在全基因组测序结果中就包含了较多具有治疗指导价值的遗传标记[40]。针对全基因组测序中覆盖的具有治疗指导意义的变异位点的临床报告制定相应的指南,也有助于推动全基因组测序技术的临床应用。
如同基因往往具有多效性,基因变异也同样可以具有多效性。例如,GBA基因的致病变异不仅可能与戈谢病(葡糖脑苷脂病)相关,其杂合子还可能提高患早发性帕金森病的风险。因此,在具体临床案例的分析中,从不同的角度看,同一个基因变异的不同效应可能对应不同的临床分级。这种情况增加了基因变异临床分级的复杂度。
由于遗传病表型的多样性及医生对此类疾病认识的相对不足,临床表型采集难免有不足之处,这些都会对临床工作中进行基因变异的临床意义分级造成影响。本方案在临床实践过程中必然会存在一些因素使得分级存在一定的主观性,但是这并不影响本方案能够使读者更好地理解遗传检测报告。同时,本方案的实施也有助于提高广大临床工作者对临床表型采集的重视程度,促进临床-实验室间的沟通,不断提高临床意义分级的合理性,从而形成一个良性循环,最终有利于基因检测报告的解读及遗传咨询。
此外,基因检测机构要标准化、规范化,要有严格的质控标准,并且要不断更新扩大基因谱检测范围,从而发现更多致病性基因,为临床提供帮助。临床医师也要熟悉和掌握基因检测相关知识,提高解读基因报告的能力与水平。
执笔:
蔡春泉 天津市儿童医院(天津大学儿童医院),天津市儿科研究所,天津市儿童出生缺陷防治重点实验室
喻长顺 天津金域医学检验实验室,广州医科大学金域检验学院
舒剑波 天津市儿童医院(天津大学儿童医院),天津市儿科研究所,天津市儿童出生缺陷防治重点实验室
李 光 天津医科大学基础医学院遗传学系
参与本建议制定的单位及人员(按单位和姓氏拼音排序):
天津大学儿童医院(陈悦,刘楠,吕玲,王萍,徐晓薇,张玉琴)
天津金域医学检验实验室(韩晓雪,刘洪洲,刘晴晴)
天津市安定医院(李洁)
天津市第二人民医院(李颖)
天津市第三中心医院(李涛,王凤梅)
天津市第五中心医院(刘晓智,姚玲)
天津市第一中心医院(刘丽,徐凤琴,张美姿,赵明峰)
天津市妇女儿童保健中心(冯树人,刘霞,辛力)
天津市海河医院(陈怀永,吴琦)
天津市环湖医院(安中平,程秀丽,李庆国,闫华,岳伟)
天津市人民医院(董昭樱,王辉,张诗武)
天津市天津医院(张春智)
天津市胸科医院(陈庆良,付博,高静,姜楠,孙大强,徐美林,张颖)
天津市眼科医院(李津,李轩,史学锋)
天津市中西医结合医院(崔云峰,王玉水)
天津市中心妇产科医院(常颖,罗海宁,任晨春,田秀英,张玥)
天津市中医药研究院附属医院(刘鹏飞)
天津医科大学第二医院(刘长山,王建梅,王雪艳)
天津医科大学基础医学院遗传学系(李卫东,时文涛,王峰)
天津医科大学眼科医院(魏瑞华)
天津医科大学肿瘤医院(任丽,于津浦,赵海丰)
天津医科大学总医院(姜丽红,李增彦,史云芳,张颖,郑荣秀)
天津中医药大学第一附属医院(夏天)
