6%苯唑草酮·唑啉草酯颗粒剂高效液相色谱分析
2021-07-08李彦飞冯泽腾张小军
李彦飞,冯泽腾,张小军
(中农立华生物科技股份有限公司,北京 100052)
苯唑草酮(topramezone),化学名称为[3-(4,5-二氢-1,2-噁唑-3-基)-4-甲磺酰基-2-甲基苯基](5-羟基-1-甲基吡唑-4-基)甲酮,是巴斯夫开发的一种苯甲酯吡唑酮类除草剂。该剂主要抑制植物体内对羟基苯基丙酮酸酯双氧化酶(HPPD)活性,并对耐三嗪类、草甘膦、ALS抑制剂和ACCase抑制剂的杂草有良好的防除效果,能有效防除玉米田阔叶杂草及禾本科杂草,但对莎草科杂草效果较差[1-5]。唑啉草酯(pinoxaden),化学名称为8-(2,6-二乙基-4-甲基苯基)-1,2,4,5-四氢-7-氧-7H-吡唑[1,2-d][1,4,5]氧二氮-9-基-2,2-二甲基丙酸酯,是先正达开发的一种苯基吡唑啉类除草剂。其可抑制乙酰辅酶A羧化酶的活性,阻碍脂肪酸的生物合成和细胞膜形成,导致杂草生长停止并最终死亡。唑啉草酯为内吸传导型、选择性禾本科杂草除草剂,广谱,高效,被茎叶吸收[6-9]。
当前国内有关苯唑草酮和唑啉草酯的复配及混剂分析方法尚无报道。基于此,本文研究了用高效液相色谱法同时检测6%苯唑草酮·唑啉草酯颗粒剂中的苯唑草酮和唑啉草酯的方法,旨在为该混配制剂生产的质量控制和标准制定提供技术支撑。
1 试验部分
1.1 仪器与试剂
1220型高效液相色谱仪配紫外检测器和色谱工作站(美国Agilent);TU-1901型紫外可见分光光度计(北京普析通用仪器有限责任公司);AL204型电子分析天平(梅特勒-托利多仪器(上海)有限公司,精度为0.000 1 g)。
乙腈(色谱纯,Thermofisher);冰乙酸(分析纯,国药集团化学试剂有限公司);新蒸二次蒸馏水;苯唑草酮标样(质量分数≥99%,北京勤诚亦信科技开发有限公司提供);唑啉草酯标样(质量分数≥99%,北京勤诚亦信科技开发有限公司提供);6%苯唑草酮·唑啉草酯颗粒剂,自制。
1.2 色谱条件
液相色谱柱:Agilent ZORBAX SB-C18不锈钢柱(250 mm×4.6 mm,内装5 μm C18填充物);流动相:乙腈-1%冰乙酸水溶液(体积比55∶45);流量:0.8 mL/min;柱温:室温;检测波长:254 nm;进样体积:5.0 μL。在上述色谱操作条件下,唑啉草酯和苯唑草酮的保留时间分别约为3.5、15.7 min。标样及样品的高效液相色谱图分别见图1、2。
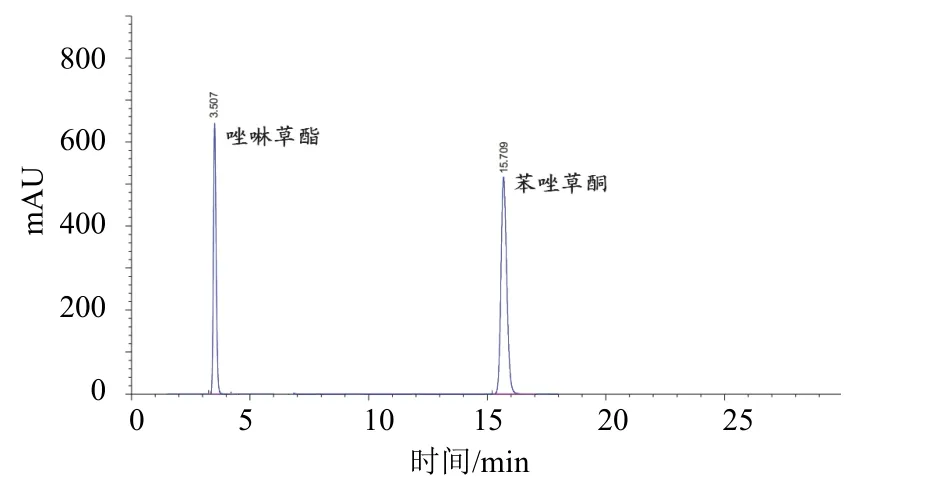
图1 唑啉草酯(进样浓度700 mg/L)及苯唑草酮(进样浓度300 mg/L)标样的高效液相色谱图
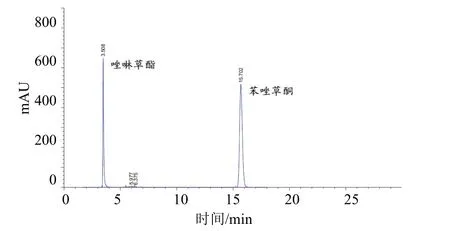
图2 6%苯唑草酮·唑啉草酯颗粒剂的高效液相色谱图
1.3 溶液的配制
1.3.1 标样溶液的配制
称取0.03 g苯唑草酮标样(精确至0.000 02 g),0.06 g唑啉草酯标样(精确至0.000 2 g),置于100 mL容量瓶中,用乙腈溶解并定容至刻度,摇匀,脱气后备用。
1.3.2 试样溶液的配制
称取1.5 g 6%苯唑草酮·唑啉草酯颗粒剂(预先将颗粒剂粉碎成末,精确至0.000 2 g),置于100 mL容量瓶中,用乙腈溶解并定容至刻度,摇匀,脱气后备用。
1.4 测定
待仪器基线稳定后,按照以上色谱条件连续注入数针标样溶液,直到相邻2针标样响应值的相对变化小于1.5%后,方可按照标样溶液、试样溶液、试样溶液、标样溶液的次序进行测定。
1.5 计算
试样中苯唑草酮(或唑啉草酯)的质量分数X(%)按下式计算:
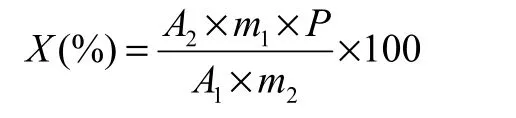
式中:A1为标样溶液中苯唑草酮(或唑啉草酯)平均峰面积值;A2为试样溶液中苯唑草酮(或唑啉草酯)平均峰面积值;m1为苯唑草酮(或唑啉草酯)标样的质量(g);m2为试样的质量(g);P为标样中苯唑草酮(或唑啉草酯)的质量分数(%)
2 结果与讨论
2.1 流动相的选择
以不同体系及同一体系中不同比例的2个物质作为流动相时,苯唑草酮和唑啉草酯的出峰时间及峰形均有较大变化。当采用乙腈-水作为流动相时,峰型明显优于甲醇-水作为流动相,且加入1%乙酸后峰型和分离度变得更好。当流动相中乙腈比例太高时,唑啉草酯出峰时间过早,经过反复测试,在流速0.8 mL/min,乙腈-1%乙酸水溶液的体积比为55∶45时,苯唑草酮和唑啉草酯的出峰时间相对适中,且峰型尖锐对称无拖尾,分离效果好。
2.2 检测波长的选择
利用TU-1901型普析通用紫外可见分光光度计对苯唑草酮和唑啉草酯标样溶液进行全波长紫外扫描,得到苯唑草酮或唑啉草酯的最大吸收波长与相关浓度响应值的紫外吸收光谱图(图3)。经验证,苯唑草酮和唑啉草酯在波长254 nm均有较大紫外吸收,且能够得到较好的峰型同时避免杂质的干扰。因此,最后选定本分析方法的检测波长为254 nm。
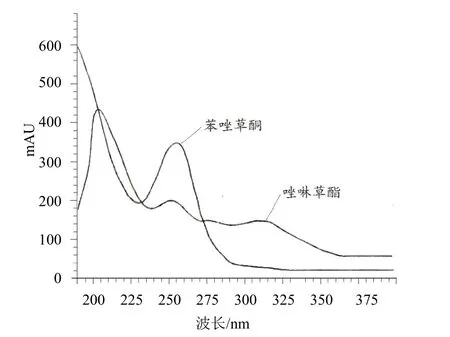
图3 苯唑草酮和唑啉草酯的紫外吸收谱图
2.3 分析方法的线性相关性
分别称取5个不同质量的苯唑草酮和唑啉草酯标样于5个100 mL的容量瓶中,配制成一系列标准溶液,按照本方法所确定的液谱条件进行进样测定,结果见表1。以苯唑草酮(或唑啉草酯)的进样质量(x)为横坐标,以苯唑草酮(或唑啉草酯)的峰面积(Y)为纵坐标做图,得苯唑草酮(或唑啉草酯)的线性相关曲线。其线性方程分别为y苯唑草酮=6 078.9x-48.218,y唑啉草酯=1 022.1x-3.031,相关系数分别为r2苯唑草酮=0.999 8,r2唑啉草酯=0.999 6,说明该方法在测定范围内呈现良好的线性关系。
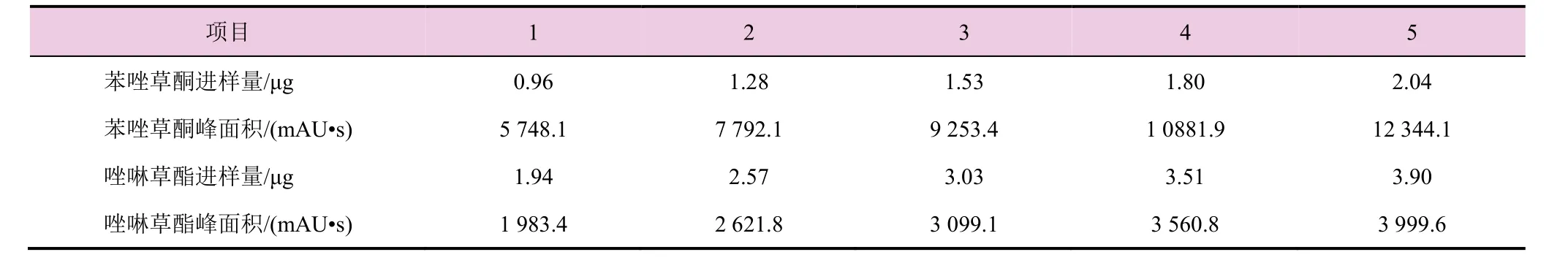
表1 分析方法的线性相关性试验结果
2.4 方法的精密度
从同一批6%的苯唑草酮·唑啉草酯颗粒剂样品中准确连续称取5个试样,在上述操作条件下进行分析,试验结果见表2。苯唑草酮和唑啉草酯标准偏差均为0.012,变异系数分别为0.59%、0.29%。由此可见,该方法的精密度能很好地满足工业分析的定量要求。

表2 分析方法的精密度试验结果
2.5 方法的准确度
准确称取5个6%苯唑草酮·唑啉草酯颗粒剂试样,加入一定量的苯唑草酮和唑啉草酯标样,按1.2节的液相色谱条件进行测定,并计算回收率,结果见表3。苯唑草酮(99.3%)和唑啉草酯(99.1%)的平均回收率均较高,符合分析标准。
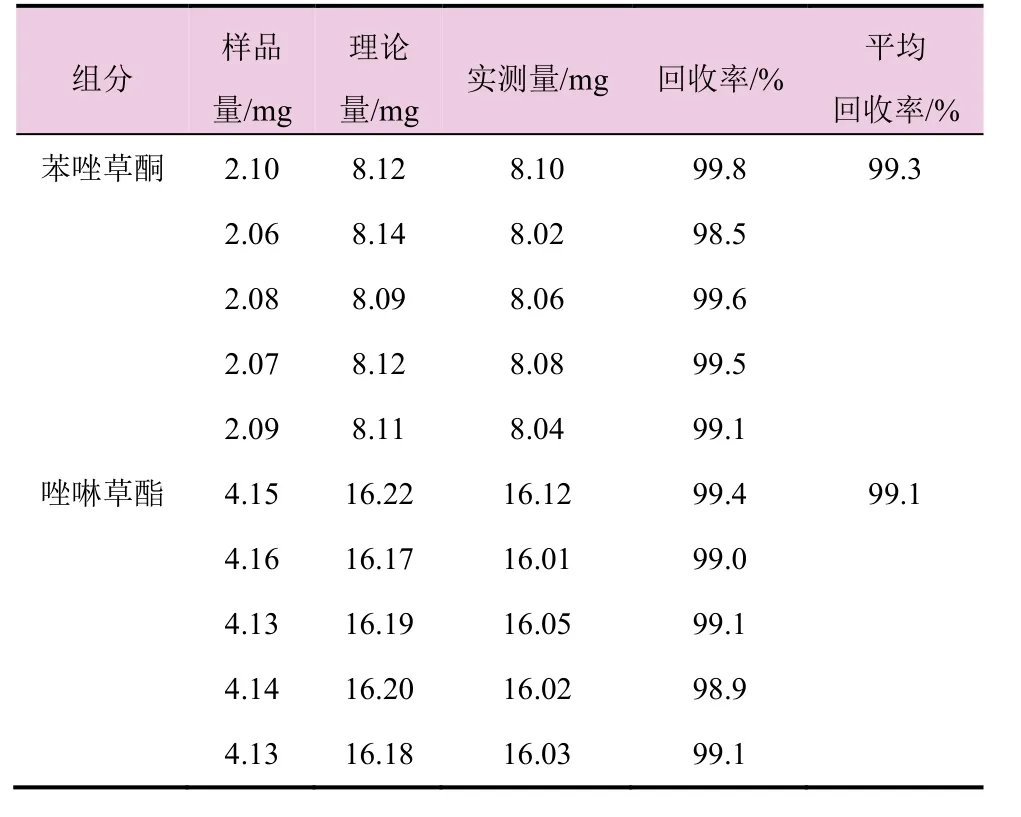
表3 分析方法的准确度试验结果
3 结 论
本文建立了用高效液相色谱法同时测定苯唑草酮和唑啉草酯混配制剂中的有效成分的定量分析方法。该方法分离效果良好,准确度和精密度均较高,且简单高效易操作,可用于苯唑草酮和唑啉草酯复配制剂的质量控制,具有很强的可行性和实际应用价值。
