抑郁、焦虑状态人群的肠道菌群构成
2021-07-03莫瀚钧柳理娜劳力敏江孙芳
莫瀚钧, 郎 林, 柳理娜, 劳力敏, 江孙芳,2*
1. 复旦大学附属中山医院全科医学科,上海 200032 2. 复旦大学附属中山医院健康管理中心,上海 200032
抑郁症和焦虑症是常见的精神心理障碍性疾病,给全世界的患者及其家庭带来巨大的经济社会负担[1-2]。其中,抑郁症是一种受基因、环境等因素影响的精神疾病,以心境低落和兴趣减少为主要的临床特征[3]。据世界卫生组织(WHO)预测,到2020年,抑郁症将成为全球成年人致残致死的首位原因[4]。而焦虑症是一种不愉快的情绪体验,表现为内心紧张,因预计某种不利情况将要出现,进而产生持续、强烈的担忧和恐惧。研究[5]表明,抑郁症患者常合并广泛性焦虑障碍,患病率为20.5%~38.0%。因此,在精神疾病负担日益增长的今天,对抑郁和焦虑症的发病机制进行研究尤为重要。
人体肠道内含有大量的微生物,其基因总数是人类自身的100多倍,因而被称为“第二基因库”,对维持机体健康发挥着重要作用。最新的多项研究[6-8]表明,肠道菌群的紊乱与焦虑、抑郁等多种精神疾病有关。Bravo等[9]发现,对焦虑、抑郁模型小鼠行迷走神经切断术后,小鼠原有的焦虑、抑郁样行为消失,提示迷走神经在肠-脑交流中的重要作用。此外,中枢神经GABA能系统的异常也被认为与焦虑、抑郁相关。逐渐形成的微生物群假说认为,微生物-肠-脑轴(microbiota-gut-brain axis, MGBA)的双向交流系统能显著影响大脑的神经生化和有关表型[10]。
目前关于肠道菌群与抑郁或焦虑的研究不在少数,然而在确诊抑郁症、焦虑障碍之前,患者往往处于抑郁、焦虑状态。对于这部分患者的肠道菌群是否已经发生了紊乱,目前缺乏相关研究证实。此外,临床上有相当一部分患者存在焦虑和抑郁的共病状态。抑郁和焦虑患者是否具有共同的肠道菌群组成,或是两者各有特异的微生物群特征,仍没有定论。因此,本研究拟采用16S rRNA测序的方法,探讨抑郁和焦虑状态人群与健康人群在肠道菌群构成上的差异,为后续进一步探索菌群异常在神经精神疾病发展中的机制奠定基础。
1 资料与方法
1.1 一般资料 选取2017年5月至2018年4月复旦大学附属中山医院全科门诊及心理科门诊收治的237例就诊者。纳入标准:年龄18~80岁;无其他精神疾病病史;经解释后,自愿参加本研究。排除标准:近2个月内使用过抗生素或者微生物调节剂;合并严重躯体疾病或有明显胃肠道症状;合并严重认知功能障碍,不能配合完成问卷调查; 妊娠、哺乳的女性。本研究通过复旦大学附属中山医院伦理委员会批准,所有患者均知情并签署知情同意书。
所有受试者分别接受抑郁症筛查量表(patien health questionnare,PHQ-9)和广泛性焦虑障碍(generalized anxiexy disorde-7, GAD-7)量表评估,以PHQ-9评分≥5分或GAD-7量表评分≥5分为研究组(n=117),PHQ-9评分<5分且GAD-7<5分为对照组(n=120)。根据是否存在抑郁合并焦虑,将研究组分为单纯抑郁组(n=38)、单纯焦虑组(n=36)和抑郁合并焦虑组(n=43)。
1.2 量表采集 调查员与研究对象进行面对面问卷调查,由研究对象自行填写问卷。在被调查者完成问卷填写后,由调查员逐一检查问卷各个条目填写的完整性。对于勾选不规范或者漏填的条目,当场及时与调查对象进行沟通确认。
本研究抑郁症状评估采用PHQ-9量表,焦虑症状评估采用GAD-7量表。PHQ-9量表共包含9个项目,每项采取0~3分的评级,得分越高表明抑郁的症状越重[11]。GAD-7量表是评价焦虑症状的自评量表[12],一共包含7个条目,同样采取0~3分进行评级,分数越高表明焦虑的症状越重。
1.3 粪便样本采集及预处理 量表填写完成后,工作人员在当天完成受试者粪便采集,新鲜粪便标本在排便后6 h内放入-80℃冰箱。精确称取粪便标本200 mg,1份放入新的2 mL无菌离心管中,用于细菌16S rDNA提取,2份放入-80℃冻存。标本留取前1周内,受试者需保证正常饮食,无腹泻。
1.4 DNA提取及测序 采用SDS裂解液冻融法进行DNA提取,基因组DNA通过PowerMax 提取试剂盒(MoBio Laboratories)提取,储存于-20°C冰箱。采用NanoDrop 1000分光光度计(Thermo Fisher Scientific,美国)测定DNA的数量和质量,进一步做琼脂糖凝胶电泳。
电泳检测后通过带16S rDNA V3-V4可变区的引物进行PCR扩增后,加入0.85× AMPure XP Beads进行磁珠纯化。纯化过后的产物进行Nanodrop检测,按照所得到的值进行等量混合,2%琼脂糖凝胶对混样进行电泳,采用AXYGEN的胶回收试剂盒回收,用Qubit对回收的文库进行定量,用qPCR进行接头效率检测,算出实际浓度后,上机测序。
使用Illumina的Hiseq二代高通量测序仪对细菌16S rDNA序列进行测序,得到所有对象样本的肠道菌群16S rDNA V3-V4可变区的原始序列。OTU(operational taxonomic units) 分析使用Vsearch v2.4.4,包括去重复序列、聚类、去嵌合体。按照97%的相似性将样品序列聚类为OTU,稀释曲线趋于平坦,说明测序深度足够。使用默认参数挑选OTU的代表序列,基于SILVA128数据库通过VSEARCH对代表序列进行物种注释,进一步生成OTU 列表,在各个分类水平上统计各样本的群落组成。记录每个样本中所有OTU的丰度和分类,去除样本中含量低于总序列0.001%的OTU。
1.5 高级生物信息统计分析 数据分析主要使用QIIME、R软件(v3.2.0)和STAMP软件。使用QIIME软件计算OTU水平的α多样性和β多样性指数。α多样性包括Chao1、Shannon和Simpson指数等,绘制等级丰度曲线(ranked abundance cuve)、稀释曲线、α多样性指数组间差异的箱型图,来比较样本间的菌群丰度和均匀度。β多样性分析主要采取基于加权UniFrac距离的主坐标分析(PCoA)展示不同组间的菌群结构差异。
通过PERMANOVA评估微生物群落结构在组间分化的标志物,使用R包“vegan”。基于R包“VennDiagram”绘制Venn 图,将组间共有和独有的OTU可视化。使用R stats包Kruskal方法比较组间各分类水平门、纲、目、科、属的差异。
基于PICRUST方法预测菌群的功能差异。使用Metagenomic Profiles (STAMP) 软件包v2.1.3对输出文件作进一步分析。从KEGG代谢途径第3层、COG功能分类2个方面进行差异比较。
1.6 统计学处理 问卷填写完成后,采用Epidata 3.1建立与问卷内容一致的数据库,由统一培训的数据录入员完成录入,然后使用Excel将数据进行整理和编码。连续变量:年龄、体质量指数(BMI)采用单因素方差分析,PHQ-9和GAD-7分数采用Kruskal-WallisH检验。分类变量:性别、既往疾病史和吸烟情况采用Pearsonχ2检验。细菌菌门间的统计学差异,2组比较采用t检验或Mann-WhitneyU检验,4组比较采用Kruskal-Wallis检验。采用t检验和the Monte Carlo permutation检验比较组间的Unifrac距离。所有检验采取双侧,检验水准(α)为0.05。
2 结 果
2.1 一般资料分析 结果(表1)显示:除了PHQ-9和GAD-7评分外,3个疾病组和健康对照组之间在年龄、性别、BMI、既往疾病史(高血压、糖尿病、冠心病)和吸烟情况上差异无统计学意义。单纯抑郁组的PHQ-9评分为(7.35±2.6)分,单纯焦虑组GAD-7评分为(7.34±2.7)分;抑郁合并焦虑组的PHQ-9评分为(9.42±4.8)分,GAD-7评分为(10.3±4.2)分,显著高于其余各组(P<0.001)。
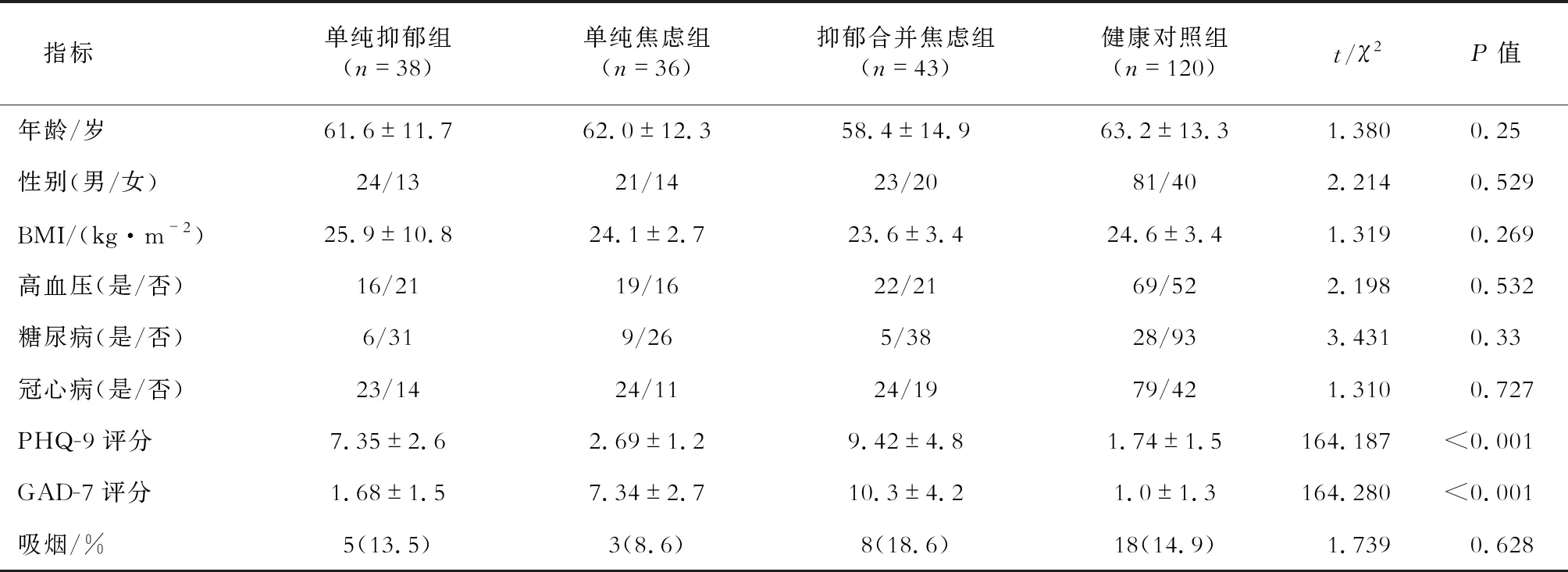
表1 4组人群的一般资料比较
2.2 抑郁与焦虑组肠道菌群结构分析 从4组人群的粪便样本中提取出16S rRNA进行PCR扩增,得到各组序列数和OTU数及组间分布的韦恩图(图1)。由韦恩图(图1B)可知,4组共有的OTU数为1 523,其中单独为抑郁和焦虑所特有的OTU数分别是41和105,抑郁合并焦虑组特有的OTU数为52,健康对照组特有的OTU为181。4组在菌群组成上有较大的重叠,组间物种区分度不理想。健康对照组的丰度高于3个疾病组;曲线下降较平缓,表明4组的群落均匀度尚可,但组间均匀度差异不大(图1C)。
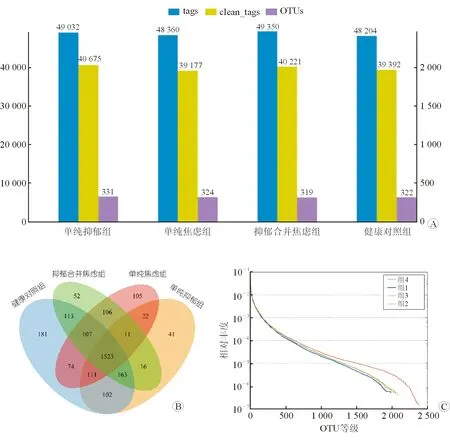
图1 4组序列数和OTU数及组间分布的韦恩图
2.3 肠道微生物群落的α和β多样性比较 比较4组的Chao1、Shanon和Simpson指数,结果(图2A)表明,3个α多样性指标在4组间差异无统计学意义。α多样性Shannon稀释曲线(图2B)显示,测序量已足够,可以反映样品中绝大多数的菌种信息。采用Weighted Unifrac PCoA的主坐标分析发现,3个实验组与健康对照组在PC1(36.02%)上差异有统计学意义(P<0.05),在PC2(13.48%)上差异无统计学意义,总体而言4组之间的β多样性亦差异无统计学意义(图2C)。

图2 实验组与健康对照组肠道微生物群落的α和β多样性比较
2.4 在门、属层面的菌群构成 结果(图3)显示:4组在门、属层面丰度排名前10位的菌群组成。从门的层面来看(图3A),不论是疾病组还是对照组,肠道菌从高到低依次是厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria)。其中,厚壁菌门和拟杆菌门的总丰度超过50%。从属的层面来看(图3B),4个组里拟杆菌属(Bacteroides)均占统治地位,剩余菌属的比例略微存在差异,但总的构成比差异不大。4组中,普氏菌属(Prevotella)的比例仅次于拟杆菌属(Bacteroidaceae),共占据了总菌群丰度的约30%。

图3 焦虑、抑郁组与健康对照组在门层面(A)和属层面(B)的菌群构成
2.5 组间菌群差异 结果(图4)显示:由图4A可见,单纯抑郁(G1)和单纯焦虑组(G2)中,酸杆菌门(Acidobacteria)和TM7菌门丰度均与健康对照组(G4)显著不同(酸杆菌门:G1vsG4,P=0.008; G2vsG4,P=0.014;TM7菌门:G1vsG4,P=0.046; G2vsG4,P=0.037)。其中,单纯抑郁组中酸杆菌门和TM7菌门均低于健康对照组,差异有统计学意义(P=0.008和P=0.046);蓝藻菌(Cyanobacteria)在单纯抑郁组的丰度也显著低于健康对照组(P=0.031)。在单纯焦虑组中,变形菌门的丰度显著低于健康对照组,差异有统计学意义(P=0.035),提示变形菌门减少可能在焦虑发病中具有重要作用。
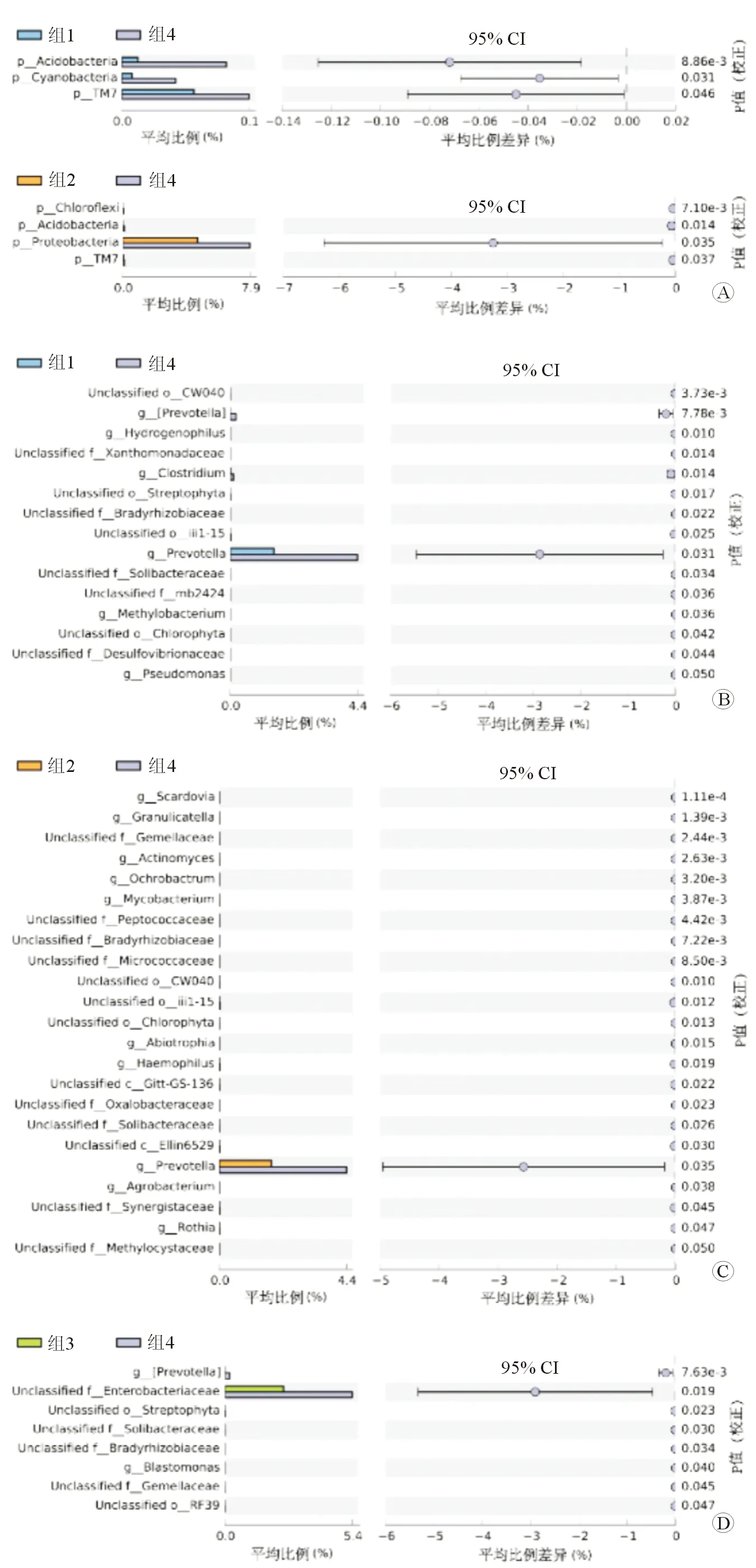
图4 各疾病组与健康对照组在门、属层面的菌群构成差异
在属的层面上(图4B~4D),不论在单纯抑郁还是单纯焦虑组中,普氏菌属的丰度均显著低于对照组,差异有统计学意义(P=0.031和P=0.035);焦虑合并抑郁组中,未分类肠杆菌属(unclassifiedEnterobacteriaceae)的丰度显著低于对照组,差异有统计学意义(P=0.019)。此外,从各组的差异菌丰度比较来看,单纯焦虑组(G2)与健康对照组(G4)差异较多,共筛选出23个差异菌属;而单纯抑郁(G1)与抑郁合并焦虑(G3)组差异菌属较为相似。
2.6 组间差异菌属分析 结果(图5)显示:与健康对照组相比,3个疾病组的差异菌属存在不同,其中单纯抑郁组、单纯焦虑组的梭菌属(clostridium)丰度均显著降低(图5A)。此外,本研究还发现霍尔德曼氏菌属(Holdemania)在单纯焦虑组(G2)中显著高于其余各组(图5B),而剩余3组中的Holdemania属丰度大致相当。柯林斯菌属(Collinsella)在单纯抑郁组和抑郁合并焦虑组,组内分布差异较大,2组菌属丰度的中位数均低于健康对照组。
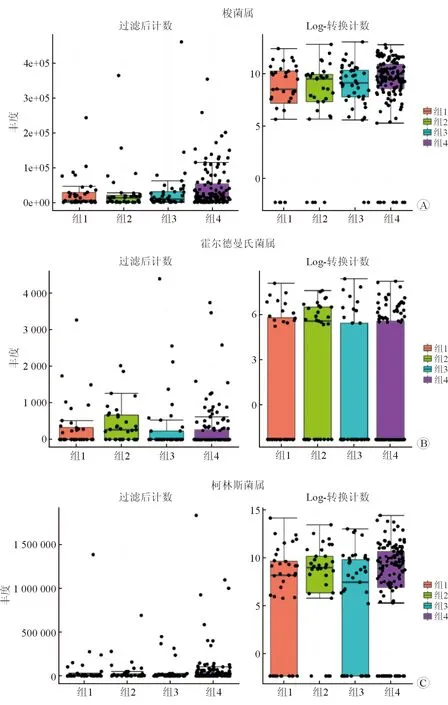
图5 4组间梭菌属(A)、霍尔德曼氏菌属(B)和柯林斯菌属(C)分析
2.6 疾病组与对照组代谢途径差异 结果(图6)显示:疾病组与对照组KEGG代谢途径差异。

图6 各疾病组与健康对照组的代谢途径的差异
单纯焦虑组的差异代谢途径较多,具体体现在花生四烯酸(arachidonic acid)代谢、氨基苯甲酸酯(aminobenzoate degradation)降解、磷酸转移酶系统(PTS)、辅酶Q的生物合成(ubiquinone biosynthesis)、不饱和脂肪酸的生物合成(unsaturated fatty acids)、脂肪酸代谢、谷胱甘肽代谢等方面显著低于对照组(P<0.05)。单纯抑郁组与抑郁合并焦虑组均有钙信号通路(calcium signaling pathway)和光合作用(photosynthesis)相关代谢通路的变化。此外,单纯抑郁组的丙酮酸代谢(pyruvate metabolism)途径高于对照组,差异有统计学意义(P=0.014)。抑郁合并焦虑组胆汁分泌(bile secretion)代谢途径显著低于对照组,差异有统计学意义(P=0.015)。
3 讨 论
本研究共纳入117例受试者为研究组,120例健康人为对照组。根据PHQ-9和GAD-7量表得分,将研究组进一步细分为单纯抑郁组、单纯焦虑组和抑郁合并焦虑组,分别与健康对照组进行比较分析。结果显示,4组在α和β多样性上存在着一定的差别,但差异均无统计学意义。
进一步的分析显示,在门的层面上,单纯抑郁组中酸杆菌门(Acidobacteria)和TM7菌门丰度构成比低于健康对照组,差异有统计学意义(P=0.008、0.046);单纯焦虑组中,变形菌门(Proteobacteria)的丰度显著低于健康对照组(P=0.035)。在属的层面上,与健康组相比,3个疾病组的差异菌存在不同,其中单纯抑郁组、单纯焦虑组和健康对照组相比梭菌属(Clostridium)均显著降低。类似的研究[13-14]也报道了抑郁患者中梭菌目(Clostridiales)和梭菌属(Clostridia)的降低,与本研究结果一致。整体上看,单纯抑郁组与抑郁合并焦虑组的差异菌属较为相似,2组普氏菌属(Prevotella)的丰度均显著降低(P=0.031、0.007)。
在代谢途径方面,本研究发现单纯焦虑组的差异代谢途径较多,单纯抑郁组与抑郁合并焦虑组均有钙信号通路和光合作用代谢通路的变化。其中,单纯抑郁组的丙酮酸代谢途径高于健康对照组,提示可能与抑郁的发病机制存在关联。荣晗等[15]通过对抑郁患者进行鸟枪法宏基因组测序,同样探讨了代谢途径的差异,发现与对照组相比,抑郁症组的泛酸酯、辅酶A合成和色氨酸代谢通路表达较高,而P53信号通路表达较少。
针对抑郁症患者的菌群改变,国内课题组已有类似的研究。Jiang等[16]率先对46例抑郁症患者和30例健康对照的粪便样本进行了16S rRNA测序,发现抑郁症患者与健康对照组相比, 拟杆菌、变形菌和放线菌增多,厚壁菌的数量减少。随后,Zheng等[17]发现,抑郁症患者的肠道菌群与健康对照组相比,α多样性无显著差异,而β多样性显著不同。具体表现为放线菌增多,而拟杆菌减少。以上实验与本研究结果存在差异,可能是因为样本量、测序平台、样本保存方法、入组患者的人口统计学、以及抗抑郁药的使用等差异造成的[18]。从这一点可以看出,目前肠道菌群的相关研究还存在较大的不一致性,可能影响结果的混杂因素也较多。因此,如何做好有关因素的控制以产出高可信度的结果,是所有研究者面临的挑战。
针对发现的菌群改变,Zheng等[17]进一步将抑郁症患者的粪便移植到了无菌小鼠,发现小鼠发生相应的菌群紊乱,并表现出抑郁样的行为。而接受健康对照组粪便的小鼠未表现出类似的行为。由此可见,肠道微生物极有可能通过多种途径,例如影响HPA轴和迷走神经,进而在焦虑、抑郁等神经精神疾病的发生发展中发挥作用。
近年来提出的“微生物-肠-脑轴(MGBA)”假说被认为是参与菌群和精神疾病互作的重要机制。根据Pu等[19]的小鼠试验结果,抑郁表型通过移植粪菌的传递很可能与MGBA有关。Bai等[20]研究了B类清道夫受体CD36,发现其在抑郁的小鼠和患者中均表达上调。敲除CD36以后,小鼠海马的NOD(nucleotide binding oligomerization domain)样受体家族3(NOD-like receptors,NLRP3)炎性体信号通路下调,盲肠细菌的α多样性增加。因此,CD36分子可能通过调节微生物-肠-炎症体-脑轴影响抑郁样行为。Deng等[21]还发现,慢性束缚应激会导致MGBA功能的异常和犬尿氨酸代谢通路的改变。随着相关研究的深入,MGBA的作用机制将变得更加清晰。
抗抑郁药也是本类研究的重要混杂因素[22-23]。Lukic'等[18]发现,与对照组相比,抗抑郁药降低了小鼠肠道菌群的丰富度并增加了β多样性。Lis'kiewicz等[24]也发现,在精神病医院进行了6周的艾司西酞普兰治疗后,粪便微生物群中的α多样性增加。然而,朱建立等[25]研究了27例首发抑郁症的患者,通过对患者的粪便进行16S rRNA测序发现,在多样性指数方面,首发抑郁症组和健康对照组的菌群差异无统计学意义。因此,尚需要进一步的研究来确定抗抑郁药对于患者菌群的影响。本研究中,患者仅表现为抑郁、焦虑状态,未达到相关用药指征,因此不存在抗抑郁药物对菌群的影响。对患者的肠道菌群进行动态随访有助于发现从健康到抑郁、焦虑发病过程中的菌群变化,并进一步发现相关机制。
有趣的是,Liu等[26]在对比了腹泻型肠易激综合征患者和抑郁症患者的粪便微生物群后,发现两者具有相似的改变,并定义了3种不同的“肠型”。由此推测,目前所发现的抑郁症较健康人群出现的特征性变化并非为抑郁症所特有,不同类型的疾病表型可能共享着某些肠道菌群的组成改变。
本研究的优势在于,共纳入了237例患者,在国内属于针对抑郁和焦虑较大样本量的研究。其次,在患者分组上,根据PHQ-9和GAD-7分数将病例组分成了单纯抑郁、单纯焦虑、抑郁合并焦虑组,很好地避免了抑郁和焦虑表型的潜在相互影响。本研究也存在一些不足:(1)在研究对象选择上,主要在全科及心理科门诊入组研究对象,就诊的患者相较于精神专科医院症状较轻,这可能在一定程度上影响了结果。(2)在焦虑和抑郁症状评估方面,选择了GAD-7和PHQ-9自评量表,均是焦虑和抑郁的筛查量表,相较于汉密尔顿量表存在一定的主观性,容易出现假阳性,给准确识别符合标准的患者带来了一定困难。此外,本研究患者的入组主要采取量表的数据,缺乏相应的临床诊断作为支持。未来的研究需要完善相关的诊断依据,进一步探讨具体菌群在抑郁、焦虑发病中的作用机制。
综上所述,本研究采用高通量测序的方法,探讨抑郁和焦虑状态的患者肠道菌群组成的差异。结果显示,单纯抑郁组、单纯焦虑组、抑郁合并焦虑组与健康对照组相比,α和β多样性未发现显著差异。在属的层面上,单纯抑郁组和单纯焦虑组中梭菌属的丰度显著降低。单纯焦虑组中霍尔德曼菌属的丰度显著高于其余各组。
