基于16S rRNA 测序探究杜洛克猪饲料利用率与肠道菌群的关系
2021-06-19田威龙司景磊莫家远刘笑笑吕冬玲李月月陈奎蓉兰干球
田威龙,司景磊,莫家远,刘笑笑,程 锋,吕冬玲,李月月,陈奎蓉,梁 靓,兰干球,梁 晶
(广西大学动物科学技术学院,广西南宁 530004)
猪在我国畜牧业养殖中占有重要地位,据《中华人民共和国2019 年国民经济和社会发展统计公报》显示,2019 年我国猪肉产量虽同比下降21.3%,但仍达4 255 万t。成本核算发现,饲料成本占整个养猪生产成本的60% 以上,同时由于猪饲料是玉米-豆粕型饲料,这在一定程度上导致了猪用粮食增加,因此提高饲料利用率是生猪产业发展的必由之路。遗传、疾病、饮食和管理等诸多因素都会影响猪的饲料利用率[1-3]。近年来随着测序技术的发展以及对猪的肠道微生物组的研究,发现肠道菌群是食物消化的重要贡献者,影响宿主的整体生理生长、免疫反应和发病机制,对猪的饲料利用率产生重要影响[4-5]。Tan 等[6]研究发现,高、低饲料转化率(FCR)长白猪肠道不同位置存在的差异微生物,揭示了与FCR 相关的潜在肠道微生物标志物。Mccormack 等[7]研究发现,饲料利用效率更高的杂交猪中有益细菌的相对丰度较高,尤其是梭菌和拟杆菌属的成员;而红球菌和红藻菌等潜在有害细菌的相对丰度较低,表明肠道菌群与饲料利用效率密切相关。因此,了解猪肠道菌群的组成和差异可为通过调节肠道微生物的群落、提高猪饲料利用效率提供一定策略。本研究以杜洛克为研究对象,探索高FCR 和低FCR 猪肠道微生物的组成和差异,为进一步挖掘与猪饲料利用率密切相关的肠道微生物提供理论基础。
1 材料与方法
1.1 动物饲养与样品收集 试验所用91 头美系纯种杜洛克公猪来自广西南宁市某核心育种场。仔猪断奶后,同时转移至相同保育条件下饲养,并在30 日龄时转移至同一育肥舍,使用奥斯本自动喂料系统对猪的采食量和体重进行测定。对所有试验猪提供相同的玉米-豆粕型基础日粮(NRC,2012),其组成及营养成分见表1。测定期间自由饮水与采食,未使用任何抗生素或药物。当达到(80±1.5)日龄时,采集个体新鲜粪便样品,放入液氮速冻后转至-80℃冰箱保存至使用。
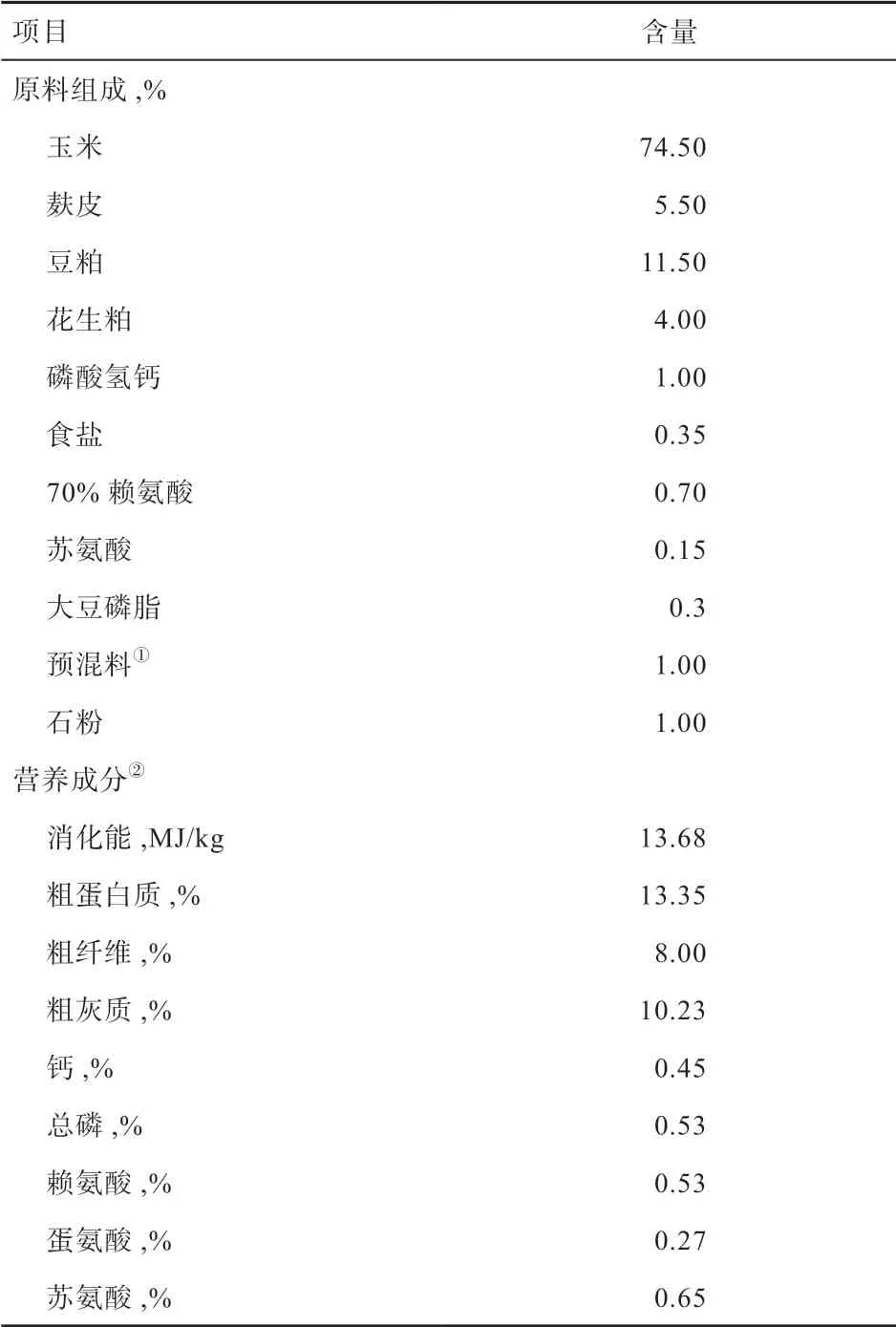
表1 育肥猪基础饲粮配方及营养成分
1.2 饲料利用率计算和分组 利用从奥斯本自动喂料系统中导出的测定期间所有猪只的采食量数据和在初测和结测时称量个体体重数据,依据《全国种猪遗传评估方案(试行)》校正公式计算出校正30~100 kg FCR 并对FCR 进行排序,将FCR 数值最低的10 只猪作为LFCR组,将FCR 数值最高的10 只猪作为HFCR 组。
1.3 粪便DNA 的提取和测序 参照 FastDNA SPIN soil kit(MP Biomedicals,Santa Ana,CA,USA)试剂盒说明书提取粪便基因组DNA。提取的DNA 使用微量紫外分光光度计(NanoDrop 2000,美国)测量DNA浓度,使用1%琼脂糖凝胶电泳检测DNA 质量。扩增细菌16S rRNA 基 V3-V4 区,引物为通用引物(F:338F,5'-ACTCCTACGGGAGGCAGCA-3';806R,5'-GGACTACHVGGGTWTCTAAT-3')。扩增条件:98℃初始变性1 min,98℃变性10 s,50℃退火30 s,72℃延伸30 s,30 个循环数,72℃延伸5 min。扩增产物经凝胶电泳检测后送上海美吉生物医药科技有限责任公司使用IlluminaMiseq 平台测序。
1.4 生物信息学分析和统计 利用 QIIME(Quantitative Insights Into Microbial Ecology)(version 1.9.1)对16S rRNA 原始数据进行质控,使用USEARCH 软件软件(version7.0)按相似度≥97% 的序列配给相同的操作分类单位(OTU);RDP Classifier 贝叶斯算法(version 2.11)对OTUs 序列分类注释;使用 R 语言对粪便菌群门和属水平的 PCoA 进行分析,并由STAMP 软件(version 2.1.3)可视化;用Mothur 软件(version 1.30.2)计算Alpha 指数,包括Sobs、Shannon、ACE、Chao1;利用PICRUSt 对OTUs 丰度标准化,并将每个标准化的OTU 对应到相应的 Greengenes,获得相应的KEGG信息,并用STAMP 对丰度化的KEGG 进行差异分析。
2 结果
2.1 样本饲料利用率统计 如表2 所示,LFCR 组的FCR为1.661~1.919,平均为1.830,说明其饲料利用效率高;HFCR 组的FCR 为2.397~2.621,平均为2.469。HFCR组和LFCR 组猪的校正30~100 kg FCR 差异极显著。

表2 HFCR 组和LFCR 组猪的FCR 表型
2.2 样本序列信息 HFCR 组获得441 444 个16S rRNA基因序列,LFCR 组获得483 951 个16S rRNA 基因序列,共925 395 个。基于每个样品的OTU 指数得到所有样品的稀释性曲线(图1),Shannon 指数和Sobs 指数曲线上升后均逐步趋于平稳,说明本试验样品测序深度能覆盖肠道菌群的大部分菌种,结果可用于后续数据分析。

图1 Shannon 指数曲线(A)Sobs 指数曲线(B)分析
2.3 Alpha 多样性分析 本研究以细菌物种的多样性指数(Shannon、Simpson)、丰富度指数(Chao1、ACE)、测序覆盖率指数(Coverage)作为猪肠道微生物Alpha 多样性的参数,结果显示,HFCR 和LFCR 组间Alpha 多样性指数均无显著性差异(表3)。

表3 杜洛克猪不同样本的Alpha 多样性
2.4 基于门和属水平的微生物群落结构组成 在门水平上,共鉴定到14 个门水平菌,其中HFCR 和LFCR 组共有门有13 个(92.86%),LFCR 组有1 个独有菌门为p__Fusobacteria(图2-A)。2 组均检测到2 个优势门(图2-C),分别为Fimicutes(HFCR 组占69.57%,LFCR 组占73.01%)和Bacteroidetes(HFCR 组占26.83%,LFCR 组占22.51%),均占据了各组总数的95%以上。在属水平上(图2-B),共鉴定到114 个属水平菌,其中HFCR 和LFCR 组共有的属有110 个,HFCR 组有1个特有的属为g_unclassified_f_Bacteroidaceae,LFCR组有3 个特有的属为g_Fusobacterium、g_1-68、g_unclassified_f_Planococcaceae。2 组均检测到4 个优势属,分别为Prevotella(HFCR 组占15.22%,LFCR 组占14.20%)、norank_f_Ruminococcaceae(HFCR 组 占11.13%,LFCR 组占9.82%)、Lactobacillus(HFCR 组占10.97%,LFCR 组占9.90%)和norank_f_Lachnospi raceae(HFCR 组占7.42%,LFCR 组占4.54%)(图2-D)。

图2 HFCR 和LFCR 组的微生物群落结构组成
2.5 基于门和属水平的微生物组成差异 利用主坐标(Principal co-ordinates analysis,PCoA)比 较HFCR和LFCR 组微生物在门和属水平上的组成,从PCoA 结果显示,在门和属水平上微生物组成均未依据FCR 表型明显分为2 组(图3)。采用LEfSe 分析比较HFCR和LFCR 组之间菌群差异性,分支图显示了HFCR 和LFCR 组中在微生物的门、纲、目、科、属分类水平上最丰富的微生物群体(图4-A),共发现23 个差异项,其中14 个在LFCR 组中富集,9 个在HFCR 组中富集,LFCR 组中的差异项多于HFCR 组。LDA 分析(图4-B)结果显示,14 个在LFCR 组中富集的菌群分别为p_Actinobacteria、g_Collinsella、f_Enterobacteriaceae、o_Enterobacteriales、g_norank_f_Enterobacteriaceae、g_1-68、g_staphylococcus、g_Staphylococcus、g_norank_o_Streptophyta、c_Chloroplast、o_Streptophyta、f_norank_Streptophyta、f_tissierellaceae、o_bacillales、Streptococcus、Christensenellaceae、Ruminococcaceae,9 个在HFCR 组中富集的菌群分别为g_unclassif ied_o_Bacteroidales、f_unclassif ied_o_Bacteroidales、g_rc4-4、p_WPS-2、g_norank_p_WPS-2、o_norank_p_WPS-2、f_norank_p_WPS-2、c_norank_p_WPS-2、g_unclassified_f_Ruminococcaceae。

图4 HFCR 和LFCR 组微生物差异分析

图3 门水平(A)和属水平(B)的 PCoA 分析
在门、属水平比较HFCR 和LFCR 组前150 种微生物类群存在显著差异的微生物,结果发现,在门水平(图5-A),WPS-2 在HFCR 组中的丰度显著高于LFCR 组;在属水平上(图5-B),unclassified_f_Ruminococcaceae、unclassified_o_Bacteroidales、norank_p_WPS-2和rc4-4在HFCR 组中的丰度显著高于LFCR 组,Collinsella、norank_f_Enterobacteriaceae、Staphylococcus、1-68和norank_o_Streptophyta在LFCR 组中的丰度显著高于HFCR 组。
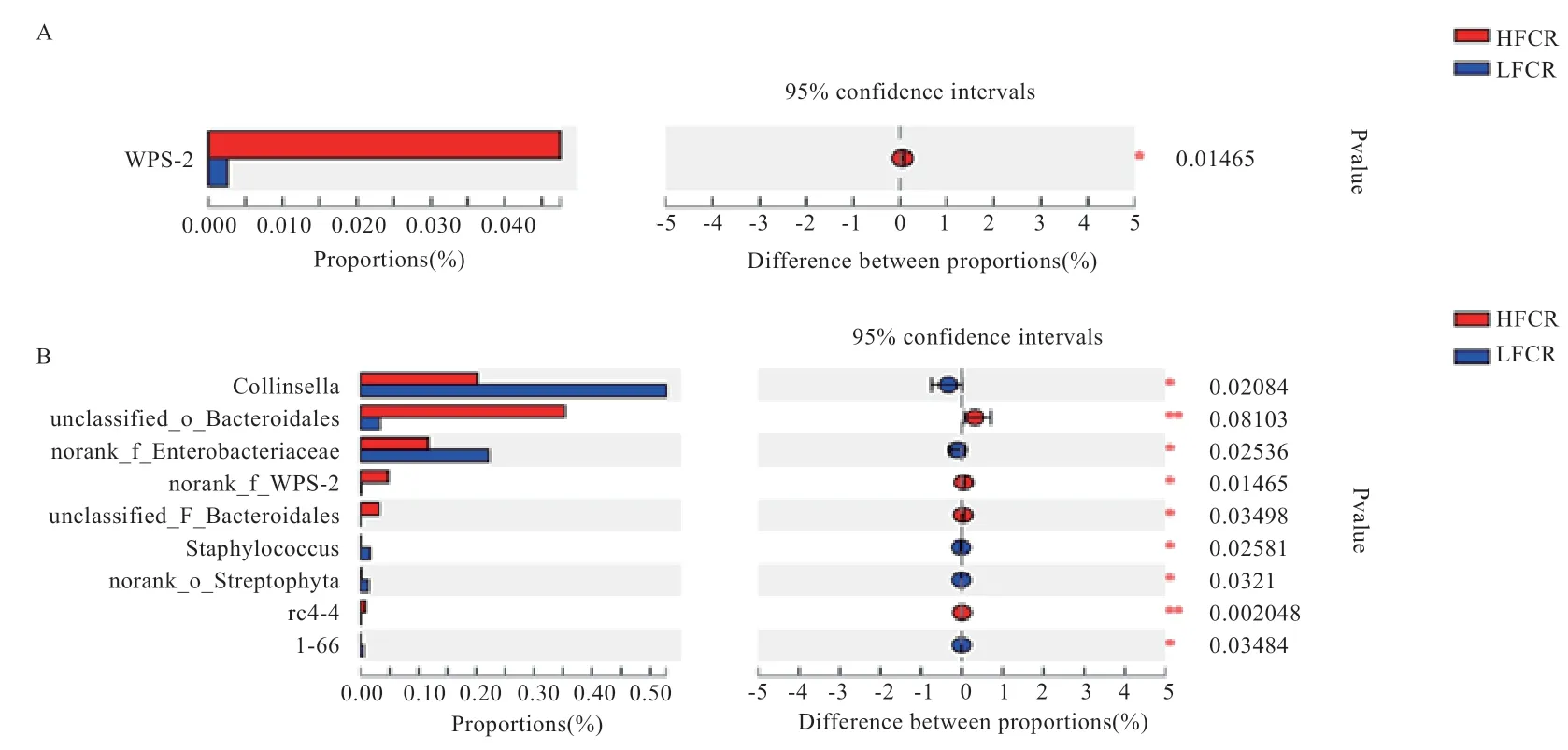
图5 在门水平(A)和属水平(B)的差异菌群
2.6 杜洛克猪肠道微生物组功能预测分析 基于PICRUSt分析平台的16S 功能预测将获得的OTU 丰度表与Greengene 数据库比对,获得相应的COG 及KO 功能信息及其丰度。结果发现,杜洛克猪肠道微生物组功能主要集中于Amino acid transport and metabolism、Carbohy drate transport and metabolism、Transcription、Cell wall/membrane/envelope biogenesis、General function prediction only 等方面(图6-A、6-B),高低组无明显差异;KEGG预测分析发现,HFCR 组和LFCR 组无显著的差异,基因主要富集Amino sugar and nucleotide sugar metabolism、purine metabolism、Glycolysis/Gluconeogenesis、Pyrimi dine metabolism 等功能通路(图6-C)。

图6 HFCR 和LFCR 组猪粪便微生物功能代谢通路分析
3 讨 论
由于我国本地猪种基本都是脂肪型猪,具有耐粗饲和抗病能力强等优点,但存在瘦肉率低、生长速度慢和饲料利用率低等问题,因此为了适应现代生猪产业的要求,我国大量引进西方商业猪种,以改良本地品种或者直接进行繁育作为商品猪,其中杜洛克通常作为最重要的终端父本使用。杜洛克在配套系中作为父本主要是其具有生长速度快、瘦肉率高、饲料利用率高等优良的生长性状,这也是杜洛克在育种工作中的主要育种目标。饲料利用率受饮食、遗传等众多因素的影响,而这些因素恰恰也是肠道菌群的重要影响因素。新一代测序技术的出现极大扩展了人们对于肠道微生物的认识,猪的肠道微生物与猪健康和生长效率之间的关系也越来越受到关注。前人发现当仔猪出生后,暴露在各种环境条件下,大量细菌开始在体内定殖,但是其肠道菌群组成会随着时间的推移而不断变化,而不是一成不变[8];尤其是在断奶后,由于饮食结构发生显著变化,肠道菌群的组成会发生很大的改变[9];而肠道微生物群和粪便微生物群之间具有密切的关系,可以通过粪便微生物的群落变化反映肠道菌群的改变[10]。所以本研究利用16S rRNA 测序技术探究粪便菌群与杜洛克饲料利用率的关系,为进一步利用肠道菌群提高饲料利用率提供参考依据。
有研究表明,肠道微生物与猪的生长性状具有一定联系[7]。本研究中试验猪均饲养在同一育肥舍,饲喂相同的育肥日粮,采取相同的饲养方式,尽可能减少外界环境因素对于肠道菌群的影响。肠道菌群呈现动态发育过程,直至育肥期才趋于稳定,故本研究测定育肥期30~100 kg 杜洛克FCR,探究此阶段高低FCR 个体间的微生物多样性差异对猪的生长性能的影响具有合理性。
本研究发现在门水平上,HFCR 组和LFCR 组中共鉴定到14 个门水平菌,其中,2 组中检测到2 个优势菌门,分别为Fimicutes 和Bacteroidetes,这与之前的报道结果类似[11];在属水平,2 组共鉴定到114 个属水平菌,其中Prevotella、norank_f_Ruminococcaceae、Lactobacillus是2 组中占据前三位的优势菌属。Pajarillo等[12]对杜洛克进行16S rRNA 高通量测序,发现普氏菌属(Prevotella)一直是其优势菌属,与本研究结果一致。研究发现普氏菌属有利于降解黏蛋白和植物性碳水化合物(如半纤维素和木聚糖),这在饲料消化中具有重要的作用[12]。本研究的优势菌属乳杆菌属(Lactobacillus)多被认为是益生菌。黄金秀等[14]研究发现,作为肠道内优势的乳杆菌之一的罗伊氏乳杆菌,可以通过重塑猪肠道菌群结构,进而影响肠道黏膜免疫功能,调节肠道生态平衡。乳酸杆菌属可在均质发酵或异源发酵中将碳水化合物转化为乳酸,或在异质发酵中将碳水化合物转化为乙酸,这2 种酸产生的酸化环境可以抑制其他微生物的生长[15],有利于维持胃肠道健康以及营养物质吸收,对于猪的生长性能也具有一定意义,也在一定程度上说明了动物的肠道菌群可以影响宿主的生长性状。
从本研究PCOA 的结果可以看出,在门和属水平上微生物组成都没有根据FCR 分为2 组,说明高低FCR杜洛克粪便微生物在菌群结构上具有一定的相似性,但是两组间仍表现出具体的差异性,这种差异性很大程度上是由于2 组个体在饲料利用率性状上的差别造成的。
LEfSe 分析显示,HFCR 组和LFCR 组中在微生物的门、纲、目、科、属分类水平上共检测到23 个差异项,可作为潜在的生物标志物,其中g_unclassified_o_Bacteroidales等9 个在HFCR 组中富集,p_Actinobacteria等14 个菌群在LFCR 组中富集。前人报道,Actinobacteria与脂肪呈正相关,暗示着Actinobacteria的富集可能有利于脂肪的沉积[16];Mccormack 等[7]研究也发现,高饲料效率猪回肠中Actinobacteria丰度是低饲料利用效率猪丰度的3 倍,说明Actinobacteria可能是潜在标志饲料利用率的菌群之一,同时也证明了粪便微生物组可在一定程度上代表肠道微生物组。属水平进一步分析发现,LFCR 组中的差异项显著多于HFCR 组且Collinsella、norank_f_Enterobacteriaceae、Staphylococcus、1-68、norank_o_Streptophyta在LFCR组中的丰度显著高于HFCR 组,与相关的报道高低组的差异菌群不符[6],这可能是由性别、年龄、品种、饮食、环境、采样方式和保存方式等诸多因素引起的。本研究是由粪便微生物代替肠道微生物,结果不同也说明粪便微生物虽然可以在一定程度上代表肠道微生物,但仍有一定差异,虽然已有研究表明肠道微生物与猪的FCR有潜在的联系,但相关菌群的功能并未研究的很透彻。此外,因为宿主本身FCR 不同导致肠道微生物组成的不同,还是因为微生物组成不同才导致FCR 不同,仍需进一步深入研究。
功能预测发现杜洛克高低FCR 组的肠道微生物组功能无显著差异,但主要集中于氨基酸运输和代谢、碳水化合物运输和代谢。小肠中的非消化性碳水化合物包括纤维素、木聚糖和抗性淀粉,可以被肠道中的微生物发酵产生挥发性脂肪酸及其衍生物,从而为微生物和宿主生长提供所需的能量[17],在育成猪配方日粮中主要成分包括富含纤维的玉米和高蛋白大豆,因此猪肠道微生物组的此种构成可能具有更高的纤维素消化利用能力;而发酵膳食纤维在肠道菌群的作用下可产生短链脂肪酸(SCFA),而SCFAs 可以减少肠道炎症,从而提高肠道的吸收能力,并提高猪的FCR,推测以氨基酸运输和代谢、碳水化合物运输和代谢为主要功能的肠道菌群是环境和饲料等多重因素导致的,可能与FCR 有关系,其机理还需要更深入研究。
总之,本研究发现高低FCR 组之间的粪便微生物组存在一定差异,支持了肠道微生物群的组成和潜在功能与FCR 相联系的假说。但这些差异被认为是微妙的,因为在所有检测到的分类单元中,差异相对较小,并且大多数确实以较低的相对丰度(<2%)出现。不过这些分类单元仍可能会影响FCR,肠道菌群内部的复杂相互作用也可能会对宿主体内稳态和FCR 产生最大影响,所以探索猪高、低FCR 肠道微生物的组成和差异,挖掘与FCR 相关的肠道菌群,进而通过饮食干预猪肠道微生物的组成,提高猪的FCR 将是一种非常有潜力的策略和研究思路。
4 结 论
本研究采用16S rRNA 技术分析杜洛克公猪高、低FCR 组的肠道微生物,揭示了与FCR 相关的复杂细菌群落。在整个菌群结构上,高低FCR 组的优势菌在类别上相同,仅在微生物的相对比例上有所差别。在高和低FCR 组中发现了与FCR 相关的潜在微生物标记物。功能预测分析发现杜洛克猪肠道微生物主要参与氨基酸运输和代谢、碳水化合物运输和代谢等营养代谢通路。本研究阐述了与猪饲料利用率相关的差异菌群,为进一步研究杜洛克猪饲料利用率与肠道微生物群的关系提供了一定的理论基础。
