磁性磺酸催化生物质基呋喃“一锅多步”合成N-取代吡咯
2021-06-16王克平郭珍燕
李 虎,王克平,杨 英,郭珍燕,韦 倩
(1.贵州大学 绿色农药与农业生物工程国家重点实验室培育基地和教育部重点实验室,贵州 贵阳 550025;2. 贵州大学 生物质资源综合利用国家地方联合工程实验室,贵州 贵阳 550025;3. 贵州大学 精细化工研究开发中心,贵州 贵阳 550025;4.贵州大学 化学与化工学院,贵州 贵阳 550025)
当前,化石能源的过度开采和使用,在满足人类社会发展的同时,也导致了环境污染、温室效应等问题。为缓解对不可再生能源的过分依赖,现有研究多聚焦于绿色可持续资源(特别是有机碳)的研发[1-3]。据估算,全球每年经光合作用生成的生物质总量约为1 700亿t,所蕴含的能量相当于世界总能耗的10倍左右;其中,碳水化合物的储量约占75%的比重[4]。生物质因含有机碳资源丰富、来源广泛、简单易得,故被认为是替代不可再生化石资源生产精细化学品和液体燃料的最佳选择[5-9]。
在适宜酸、碱、金属催化剂的作用下,生物质可经系列化学或生物催化转化路径,制得含氧化合物(如醇、有机酸、呋喃衍生物、酚类等)[10]。值得注意的是,甲壳素、壳聚糖、蛋白质等含氮生物质原料,可经系列降解并耦合胺化反应合成出含氮化合物,如丙烯酰胺、1,4-丁二胺、N-甲基吡咯烷酮、氨基酸等含氮高附加值化合物[11-13]。在众多含氮化合物中,N-杂环化合物往往表现出良好的生物活性[14-16]。特别地,吡咯衍生物作为一类重要的五元含氮杂环骨架,广泛存在于一系列商品化药物分子中(例如,用于治疗阿尔茨海默病的阿洛西坦、预防心血管疾病的阿托伐他汀等),并应用于高分子、催化剂、染料、食品、农药等领域[17]。
一般地,N-杂环化合物的合成过程往往涉及C—N和C—C键的形成(例如,胺化、环加成、缩合等反应),其合成方法或工艺往往较含氮直链分子更为复杂[16]。采用石油基化学品为原料,吡咯衍生物可经Paal-Knorr、Barton-Zard、多组分缩合或环加成等反应制得[18-19]。近年来,George W.Huber教授、Ning Yan教授、Sangho Koo教授等国内外课题组先后报道了直接催化甲壳素、虾壳、甘蔗渣等生物质原料合成吡咯衍生物;然而,所得产率往往较低(<20%),并且反应温度较高(300~600 ℃)[20-23]。转而,曹勇教授、张泽会教授、Johannes G.de Vries教授等课题组先后选用呋喃、2,5-己二酮等生物质平台小分子为原料,可在温和条件下获得较高的催化胺化活性[24-26]。值得注意的是,上述转化体系所需催化剂多为负载型贵金属或均相金属材料、催化剂,回收步骤繁琐。与之相比,非贵金属储量高、廉价易得,在催化合成生物质基吡咯衍生物方面更具开发潜能[27-28]。为克服上述催化体系中存在的不足,本文设计和制备了一种新型磁性碳基磺酸材料,并成功应用于催化生物质基2,5-二甲基呋喃与胺类化合物经“一锅多步”反应合成出一系列N-取代吡咯衍生物。同时,该催化反应体系表现出易分离回收,合成工艺绿色、可持续等特点。
1 实验部分
1.1 材料
2,5-二甲基呋喃(99%)、FeCl3·6H2O(98%)、壳聚糖(95%)、氨水(AR,25.0%~28.0%)、戊二醛(质量分数为50%的水溶液)、正硅酸四乙酯(99%)和苯胺(99%)均购于阿拉丁。
1.2 催化剂的准备
1.2.1磁性Fe3O4的制备
首先,向250 mL的圆底烧瓶中加入5.40 g FeCl3·6H2O、2.78 g FeSO4·7H2O和80 mL去离子水,于室温下快速搅拌至完全溶解。随后,用氨水调节混合液的pH值至10,并将反应体系转移至油浴锅中,加热温度和时间分别设定为80 ℃和1.5 h。反应结束、冷却到室温后,用磁铁吸附瓶壁分离出磁性固体沉淀物,并将其置于80 ℃烘箱中干燥6 h,即制得磁性Fe3O4。
1.2.2磁性Fe3O4@SiO2的制备
将上述所制1.0 g Fe3O4、20 mL去离子水、80 mL乙醇和1 mL氨水加入到250 mL的圆底烧瓶中,将反应体系磁力搅拌10 min,使颗粒均匀分散。然后,向混合液中,加入1 mL正硅酸四乙酯(TEOS),在室温下搅拌6 h,用磁铁将圆底烧瓶中的固体分离出来,得到的固体用去离子水洗涤1~2次,再用无水乙醇洗2~3次,洗净后于真空干燥箱80 ℃干燥6 h,即得Fe3O4@SiO2。
1.2.3Fe3O4@SiO2@CS-SO3H的制备
向50 mL 1%的乙酸水溶液中,加入0.5 g壳聚糖(CS),于室温下搅拌24 h,使壳聚糖溶解。然后加入1.0 g Fe3O4@SiO2,磁力搅拌10 min,使其分散均匀。随后,加入5.7 g对甲基苯磺酸,搅拌10 min后,缓慢加入1 mL戊二醛(质量分数为50%的水溶液),并继续搅拌3 h,得到凝胶状固体。过滤分离出固体,置入真空干燥箱80 ℃下干燥6 h,研磨成小颗粒后用无水乙醇洗涤3~4次至中性(pH试纸测定)。干燥后所得催化剂为Fe3O4@SiO2@CS-SO3H;为便于书写,催化剂命名为WK-8。
1.3 催化剂表征
采用Tongda TD-3500型X射线衍射仪(Cu Kα 辐射λ=0.154 056 nm)记录样品的XRD图,2θ的范围为5~80°。采用KBr 360型Nicolet红外光谱仪获得样品的红外光谱图,每次自动扫描背景,样品通过KBr压片后扫描32次,范围为400~4 000 cm-1。用热重分析法(TGA,Mettler-TGA/DSC1)测定了氮气气氛下,样品于25~800 ℃温度下的质量损失,升温速率为10 ℃/min。采用扫描电子显微镜(SEM,JSM-6700F,5 kV)观察催化剂的形貌。利用Thermo ESCALAB 250设备(Al-Kα阳极,hν=1 253.6 eV)进行样品的X射线光电子能谱(XPS)分析。
1.4 反应过程
1 mmol 2,5-二甲基呋喃、1 mmol苯胺或其他类型的芳胺(例如,对甲氧基苯胺、3-氟苯胺、3-甲氧基苯胺、间甲苯胺、邻甲苯胺、3-硝基苯胺、4-硝基苯胺、对三氟甲基苯胺)和4 mg 萘(内标)混合加入到10 mL的反应釜中,随后置入带有磁力搅拌装置的恒温油浴锅中反应1~3 h。反应结束后,将反应釜从油浴锅中取出,并立即放到自来水下冲淋,快速冷却至室温。所得的产物用4 mL的无水甲醇稀释,产物用GC(Agilent GC6890, HP-Innowax 19091 N-113,30 m×0.32 mm×0.25 μm)来定量分析。
2 结果和讨论
2.1 催化剂的结构表征
图1(a)是催化剂WK-8的FT-IR光谱图。在569 cm-1处是Fe3O4的Fe—O键振动吸收峰[29],1 531 cm-1和1 379 cm-1处分别为N—H和C—O键的振动峰,以及3 378 cm-1处是O—H的伸缩振动峰[30]。上述红外光谱结果证实CS成功地包裹在Fe3O4的纳米微球上。此外,—SO3H的特征吸收峰出现在1 180 cm-1和1 040 cm-1附近[31-33],表明磺酸基团成功地负载到Fe3O4@SiO2上。
TGA曲线如图1(b)所示。WK-8催化剂在温度为0~800 ℃的范围内发生了失重和分解,且可初步分为3个阶段。首先,在温度为0~200 ℃范围内,催化剂的质量降低了5.37%,可归结为样品中残留水分和壳聚糖上羟基的丢失[29];其次,在温度为200~500 ℃范围内,失重43.61%,可归因于有机物组分的分解[34];再次,在温度为500~800 ℃范围内,失重5.7%,主要是由于部分Fe3O4在高温下发生了分解。鉴于催化合成吡咯衍生物的体系所需反应温度不高于170 ℃,WK-8催化剂可在相应反应条件下保持良好的稳定性。
XRD曲线如图1(c)所示。2θ为30.2°、35.6°、43.3°、57.3°和62.9°处的衍射峰依次归为Fe3O4的(220)、(311)、(400)、(511)和(440)晶面,与标准Fe3O4(JCPDS数据库,190629)和γ-Fe2O3(JCPDS数据库,895894)的衍射峰完全吻合[35-42]。从XPS全谱可以看出铁、氧、碳、氮、硫、硅为WK-8催化剂的主要元素(图1(e))。催化剂WK-8的Fe2p XPS窄谱(图1(f))再次确证Fe3O4的存在:Fe2p3/2(710.9 eV)和Fe2p1/2(724.3 eV)。
由NH3-TPD图谱(图1(d))可以看出,WK-8催化剂在解吸温度为110~200 ℃、200~350 ℃和350~550 ℃附近出现了3组峰,分别对应于弱酸位点、中酸位点和强酸位点,并且测得总酸量为3.474 mmol/g。
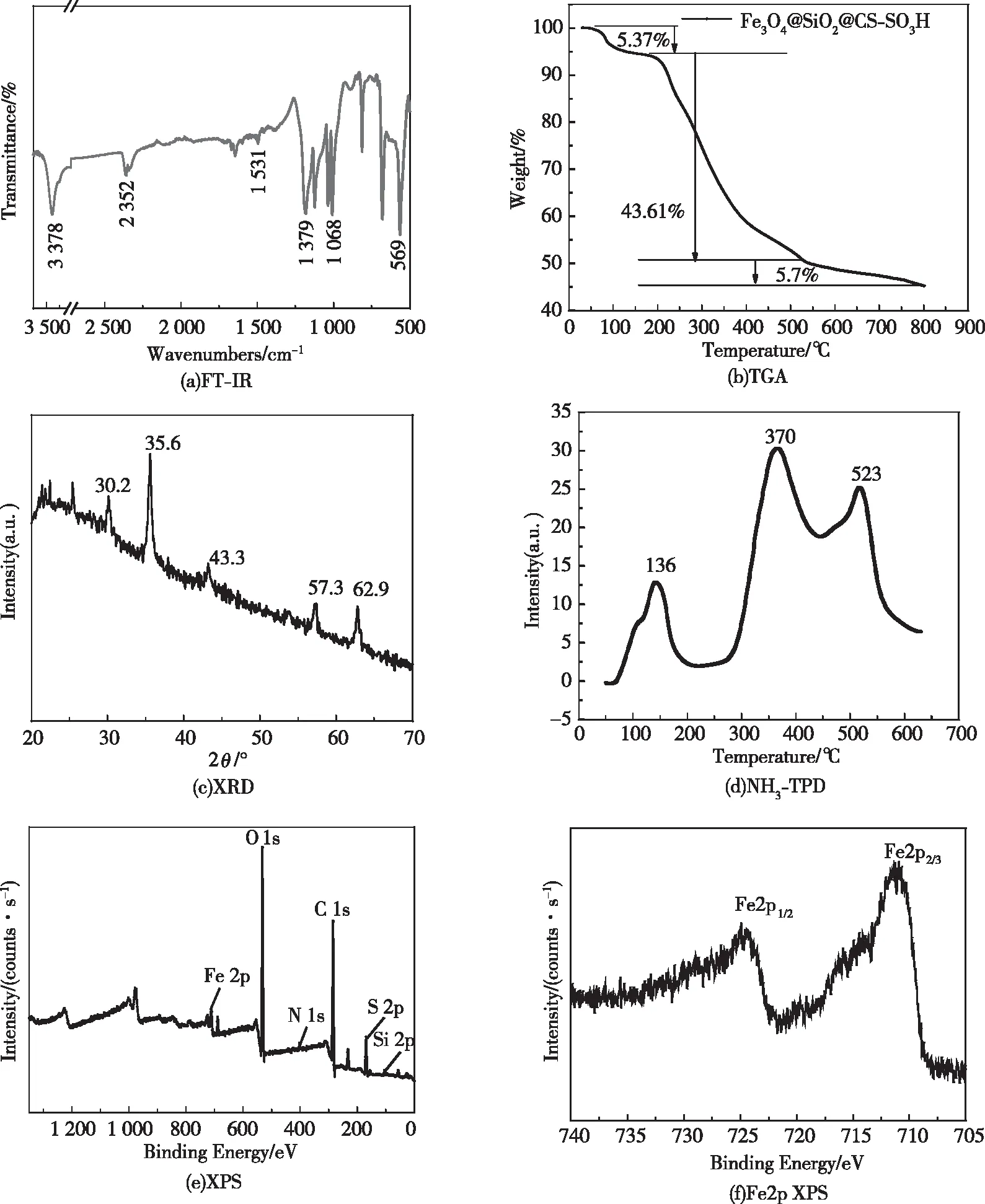
图1 WK-8表征图谱
催化剂磁性吸附分离示意如图2所示。从图2可以看出,WK-8具有磁性,用磁铁能将其快速地从体系中吸附分离出来。

图2 催化剂磁性吸附分离示意图
从WK-8的SEM扫描图(图3)观察到其表面相对无孔,且呈块状分布。

图3 催化剂WK-8的SEM图
2.2 反应条件优化
2.2.1反应温度
由图4(a)可以看出:在WK-8的催化作用下,2,5-二甲基呋喃和苯胺于110 ℃温度下反应2.5 h,所生成2,5-二甲基-N-苯基吡咯的产率仅为23.9%;当温度升至130 ℃时,产率可达75.6%。由此可见,该级联反应对温度较敏感。推测是因为转化过程可分为开环、关环、脱水等多步反应,升高温度尤其有利于反应体系开环、脱水。进一步地,反应温度由130 ℃增加到170 ℃,产率也相应提升(约13%)。鉴于后续增加幅度不大,170 ℃选为进一步优化的最佳反应温度。
2.2.2反应时间
在170 ℃反应温度下,以0.5 h为区间设置6个时间段优化反应时间。从图4(b)可以看出,时间从1 h延长到2.5 h,2,5-二甲基-N-苯基吡咯的产率相应地由64.4%增加到88.9%。鉴于酸催化2,5-二甲基呋喃开环转化为2,5-己二酮的反应过程可逆,可通过适当延长反应时间和加水促进其向正反应方向发生。值得注意的是,持续延长反应时间至3 h,2,5-二甲基-N-苯基吡咯的产率为76.9%,相比于2.5 h所得产率略有下降。可能原因是吡咯衍生物在酸性条件下不稳定,易分解转化为其他含氮化合物。因此,最佳反应时间选定为2.5 h。
2.2.3含水量
如图4(c)所示:随着含水量的增加(0~200 μL),目标产率也逐渐升高;但当加入水量超过200 μL时,所得2,5-二甲基-N-苯基吡咯的产率反而下降。这一结果很好地反映了2,5-二甲基呋喃开环合成2,5-己二酮的转化过程是可逆的;其中,水即作为反应物掺入开环过程,又作为生成物抑制脱水过程。显然,过量水虽然促进了2,5-己二酮的生成,但抑制了下游Paal-Knorr反应的发生。因此,选出一个合适的含水量对提高2,5-二甲基-N-苯基吡咯的产率极为关键,相比而言,使用200 μL去离子水最为合适。
2.2.4催化剂用量
催化剂的制备成本一般较高,少量、高效、可重复使用是衡量一种催化剂能否适用于反应体系的重要指标。在优化催化剂用量(质量分数范围为5%~20%)的实验中发现,适量增加催化剂用量,活性位点数随之增加,催化效率也明显升高。如图4(d)所示,当催化剂用量增加到质量分数15%(即14.4 mg)时,2,5-二甲基-N-苯基吡咯的产率达到最高(为88.9%)。然而,继续增加WK-8催化剂用量时,产率略有所下降(如用量为质量分数20%时,产率为76.7%)。原因可能是过量催化剂磺酸位点会诱发终产物的进一步转化或者降解反应,进而降低其收率。因此,选择14.4 mg的催化剂用量更适于本反应体系的后续优化。
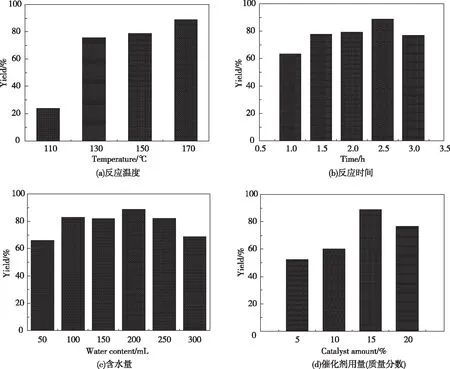
图4 2,5-二甲基呋喃与苯胺反应的单因素优化
2.2.5不同类型酸催化性能比较

表1 不同催化剂催化2,5-二甲基呋喃与苯胺的活性对比
2.2.6溶剂种类
以2,5-二甲基呋喃和苯胺的反应为模型反应,研究了9种有机溶剂对反应的影响。如表2所示,非质子极性或低极性有机溶剂如N,N-二甲基甲酰胺、二氯甲烷和环己烷,WK-8催化所得2,5-二甲基-N-苯基吡咯的产率最高只有11.4%。相反地,质子溶剂如甲醇、正丁醇、异丙醇,所得2,5-二甲基-N-苯基吡咯的产率高达77.5%~88.9%。同时,质子类有机溶剂对本体系的影响表现为,极性越强,目标产物的产率越高。可以推测,对Paal-Knorr反应而言,强极性质子溶剂更有利于质子酸在催化体系中的传输,进而极大促进反应的发生。因此,选择甲醇作为最佳有机溶剂。
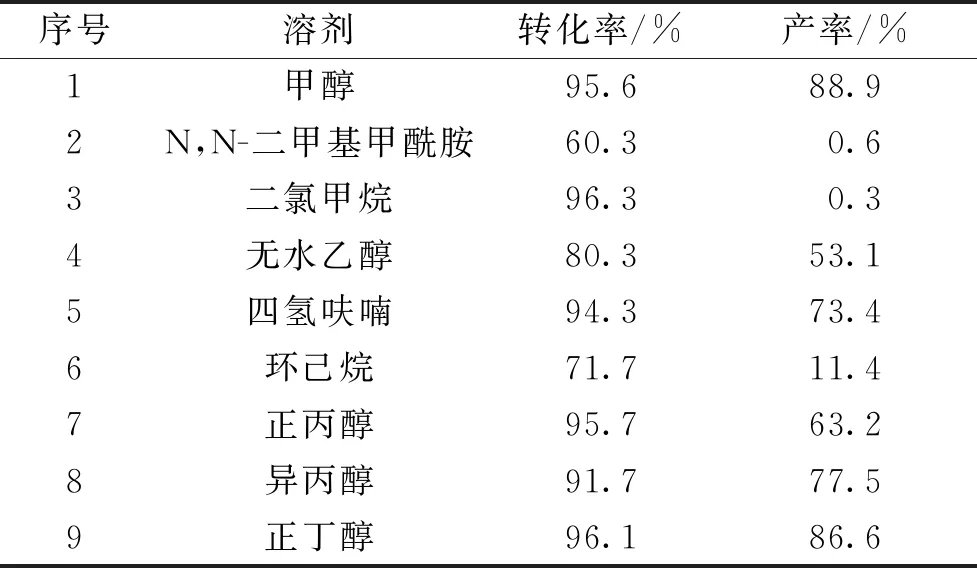
表2 溶剂对2,5-二甲基呋喃与苯胺反应的影响
2.2.7反应机理
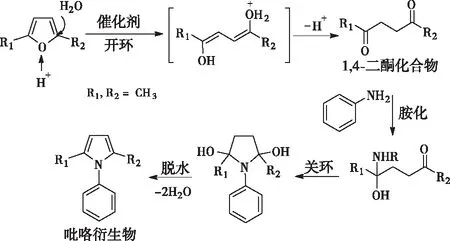
图5 催化2,5-二甲基呋喃与苯胺合成2,5-二甲基-1-苯基吡咯的机理示意图
2.3 底物拓展
基于上述最优反应条件(170 ℃、2.5 h、WK-8为催化剂),催化采用等摩尔的2,5-二甲基呋喃与不同结构的苯胺(含邻位、间位、对位取代苯胺)缩合反应制备2,5-二甲基-N-芳基吡咯,如图6所示。
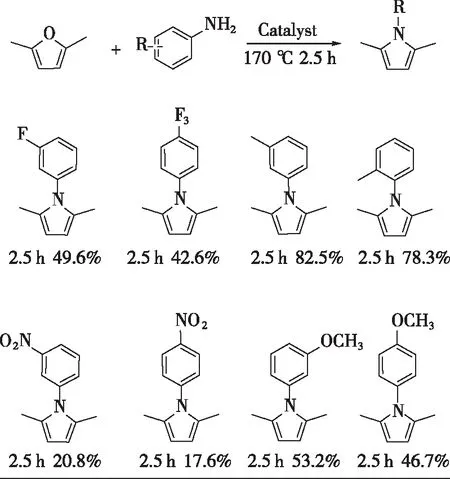
反应条件:1 mmol 2,5-二甲基呋喃,1 mmol芳胺,2 mL甲醇,200 μL H2O,14.4 mg WK-8催化剂,170 ℃,2.5 h,4 mg萘作为内标。
由图6可以看出:甲基取代基无论在苯胺邻位、间位都能选择性地与2,5-二甲基呋喃催化转化为相应的甲基取代吡咯(邻位78.3%、间位82.5%);甲氧基取代基在苯胺间位、对位的也能转化为相应的吡咯衍生物(间位53.2%、对位46.7%)。结果表明,该催化转化工艺不仅适于2,5-二甲基-1-苯基吡咯的合成,在适宜反应条件下,也可催化制备其他N-取代吡咯衍生物。
3 结论
本文研究制备了一种新型的催化性能优异的磁性碳基磺酸催化剂(WK-8),具体表现为:一方面,能有效催化生物质基2,5-二甲基呋喃与取代苯胺制备出一系列2,5-二甲基-N-芳基吡咯;另一方面,系列表征手段证实该催化剂性质稳定,在外加磁场下易于从反应体系中分离回收。控制实验证实2,5-二甲基呋喃的呋喃开环为级联反应的决速步,2,5-己二酮为反应的关键中间体。在WK-8催化剂Brnsted酸位点的作用下,水的加入能显著促进开环反应的发生,进而高效合成出目标产物2,5-二甲基-N-芳基吡咯。该磁性碳基磺酸催化剂制备工艺简单、可持续,克服了传统催化材料回收程序繁琐、制备成本高等问题。
