光谱学与分子对接结合解释生物大分子与配体间作用机制的研究进展
2021-06-11周玉玳戴双雄蔡政旭石建兵董宇平
周玉玳,戴双雄,佟 斌,蔡政旭,石建兵,董宇平
北京理工大学材料学院结构可控先进功能材料与绿色应用北京市重点实验室,北京 100081
1 引言
分子对接,通常被用来计算生物大分子和小分子配体之间可能存在的各种结合构象,构筑一个受体-配体的构象集,并预测结合力的强弱及种类,根据构象和能量的匹配排名打分,到目前为止已经被使用几十年[1-6]。1894年,Fischer[7]最早提出“锁钥模型”用于解释受体-配体间的相互作用,即受体和配体间通过几何互补和能量匹配识别彼此。这种模型认为配体和受体均为刚性结构,但是无法解释一些受体和配体结合后构象明显改变的现象。1958年,Koshland[8]在“锁钥模型”的基础上,提出了酶促反应的“诱导契合”学说,该理论表明酶和底物之间的反应,酶活性位点的空间构型受配体影响而改变,认为配体和蛋白质都是柔性结构。诱导契合理论为分子对接提供了理论参考,将受体和配体考虑为柔性结构使对接结果更加准确。分子对接的方法通常分为三类:刚性对接、半柔性对接和柔性对接。刚性对接将受体和配体均考虑为刚性,降低了对接难度并减少了计算量,适用于对结果精度要求不高或结构较为庞大的体系,如蛋白-蛋白及蛋白-核酸间的对接。半柔性对接目前使用比较广泛,将大分子受体视为刚性,小分子在结合位点处构象可以发生改变。柔性对接认为配体和受体的构象可以自由改变,计算耗时较长,多用于精确考虑结合相互作用。比较常用的对接软件有DOCK、AutoDock、AutoDock Vina、GOLD、GLIDE、SYBYL、Discovery Studio等。
分子对接可以评估配体-靶标的互补性,是小分子药物发现中至关重要的一步,结合高通量筛选技术,在计算机辅助药物设计(CADD)方面已有广泛的应用,如抗癌症药物筛选[9]、酶抑制剂筛选[10]、抗病毒药物筛选[11,12]等。由于其打分函数的优势,这种方法能够显著提高药物筛选的成功率,在实际的药物合成前节约时间及成本。除了传统的小分子药物筛选,已经有文献报道将分子对接用于大分子受体与小分子配体结合的机理解释,更多的关注小分子在受体上的结合位置及相互作用的情况[13-15]。在机理解释方面,利用对接可以得到受体-配体间结合位点、结合位点处的配体构象、配体与其周围的氨基酸残基相互作用类型、距离及结合能打分评估结果等信息。生物大分子受体与配体间的相互作用类型通常包括:静电作用、氢键作用、范德华力作用及疏水相互作用等。
传统的生物学表征手段,通常有耗时、价格昂贵、技术壁垒高及灵敏度不够等缺点,因此,越来越多的工作中使用光谱学手段来表征大分子受体与小分子配体结合的相互作用,如紫外-可见光谱(UV-Vis光谱)、荧光光谱(FL光谱)、圆二色光谱(CD光谱)、傅里叶红外光谱(FT-IR光谱)及三维荧光光谱(3D荧光光谱)等。将光谱学手段即实验结果与分子对接结合,能够更清楚地在分子层面上解释大分子与小分子的结合相互作用。例如,Liu等[16]研究了三种抗癌药物氧化白藜芦醇(OXY)、白皮杉醇(PIC)和米托蒽醌(MIT)与人血清白蛋白(HSA)的相互作用机制,荧光猝灭结果显示,三种抗癌药物与HSA蛋白作用强弱顺序为:OXY>PIC>MIT,分子对接计算的结合能的数值大小对比与猝灭结果一致,此外,计算出这三种药物的结合位点均在BSA蛋白的子域 IIA处,利用对接分析了结合口袋附近的氨基酸残基,以及氨基酸残基与药物小分子形成氢键作用的距离等。
目前已有多篇综述总结了小分子对生物大分子的检测[17,18],但是这些研究大部分侧重于光学现象和性质,也有一些文章综述了分子对接在药物设计方面的应用[19],但还没有专门的综述报道分子对接与光谱学结合用于配体与生物大分子结合机理的解释。因此,本文针对这一领域,将近年来的成果进行了较全面的综述,并对其进行客观的评论及前景展望。
2 生物大分子与配体结合的相互作用
2.1 蛋白质-配体
2.1.1UV-Vis方法

Ma等[23]在研究十二烷基硫酸钠(SDS)与胰蛋白酶相互作用时,使用了UV-Vis手段进行表征(图1A)。向胰蛋白酶溶液中加入SDS的过程中,280 nm处的紫外吸收峰强度发生下降,推测是由于SDS与芳香性氨基酸残基Tyr和Trp之间相互作用。分子对接得到的相互作用的二维图显示SDS与Trp215和Tyr228间相互作用,与UV-Vis光谱结果一致。Menezes等[24]研究了牛血清白蛋白(BSA)和人转铁蛋白(HTF)与四种糠醛衍生物的相互作用,BSA蛋白的194 nm及275 nm处的吸收峰发生改变,揭示了BSA蛋白构象的转变并确认了BSA与5-甲基-2-呋喃丙烯酸(Fur4)化合物相互作用(图1B)。HTF蛋白的变化与BSA蛋白相似。
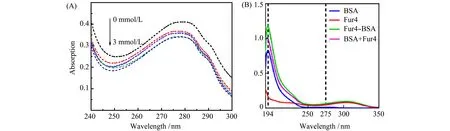
图1 (A)SDS与胰蛋白酶的UV-Vis光谱[23];(B)BSA蛋白、醛糠衍生物Fur4、BSA与Fur4混合物Fur4-BSA的UV-Vis光谱,BSA+Fur4为BSA与Fur4曲线的加和[24]
2.1.2FL方法
FL光谱是研究蛋白质-配体复合物常用的光学方法,一些蛋白质结构的变化可以通过检测发射峰的变化来判断[25,26]。以下两种情况下,常使用荧光光谱分析蛋白质与小分子间的相互作用,一是利用同步荧光光谱分析蛋白残基附近微环境的改变,二是结合Stern-Volmer方程(式1),进行热力学分析[16,27]。小分子使蛋白质荧光猝灭机制可以分为两种:静态猝灭和动态猝灭,使用Stern-Volmer方程可以区分这两种猝灭类型。温度越高,猝灭常数Ksv的数值越小,蛋白质与小分子的结合符合静态猝灭机理[28]。对于静态猝灭,利用公式(2)[29]可计算出结合常数K和结合位点数n。
F0/F=1+Kqτ0[Q]=1+Ksv[Q]
(1)
(2)
(3)
ΔG=ΔH-TΔS
(4)
式中,Kq是猝灭速率常数,τ0是不含猝灭剂的生物大分子的平均寿命,[Q]代表猝灭剂分子的浓度。
对于普通的UV-Vis光谱或FL光谱,Tyr和Trp残基的吸收或发射相互重叠难以区分。同步荧光光谱通过同时扫描激发和发射波长,可以获得发色团周围的微环境信息,通常用来表征蛋白质的构象转变。将同步荧光光谱的激发波长与发射波长的固定间隔Δλ分别固定为15或60 nm,能够分别提供关于Tyr和Trp残基的结构信息。若同步光谱的最大发射峰发生红移,说明在该波长间隔下测得的Tyr或Trp残基附近的疏水性降低,肽链的伸展程度增加。相反,如果发生蓝移,则表明环境的疏水性增加,大分子更趋向于折叠态。
Kong等[30]的研究中探讨了木糖醇XY与α乳清蛋白(α-La蛋白)等组分的相互作用,结果发现:当Δλ= 15 nm时,α-La蛋白最大发射波长蓝移3.9 nm,表明Tyr残基附近的疏水性增加。而Δλ=60 nm时,没有明显的变化,Trp残基附近的极性保持不变,说明木糖醇XY与α-La蛋白相互作用时改变了蛋白质的构象,相互作用位点更靠近Tyr残基。分子对接结果显示XY通过氢键作用结合到芳香性团簇Ⅱ附近,与XY形成氢键作用的氨基酸包括Gln 54、Tyr 103、Thr 33、Glu 49、Leu 105和Ala 106,距离分别为0.218、0.225、0.119、0.227、0.220/0.225和0.257 nm。
Wang等[31]研究了碘对胰蛋白酶和胃蛋白酶的活性和构象结构的影响。从同步荧光光谱的结果分析可知,I2诱导了胃蛋白酶和胰蛋白酶四级结构的改变并导致Tyr和Trp残基处微环境的改变(图2)。除此之外,作者利用Stern-Volmer方程(式1),将没有小分子存在时的荧光强度F0、有小分子存在时的荧光强度F等参数代入到方程中,判断出猝灭机制主要为静态猝灭,I2与胰蛋白酶结合的Ksv和Kq值大于I2与胃蛋白酶,表明I2更容易猝灭胰蛋白酶的荧光。根据公式(2)计算出结合位点数n及结合常数K,比较K值大小可知胰蛋白酶更容易与I2结合。结合过程中涉及的热力学参数通过公式(3)和(4)可计算得到,负值的ΔG表明结合是自发进行的。通过对I2在两种蛋白酶结合位点处的对接结果分析,I2更靠近胰蛋白酶的催化活性中心,与实验结果一致。分子对接得到的I2与胰蛋白酶、胃蛋白酶的结合能差别较小,分别为-3.83和-3.81 kcal/mol,与荧光光谱的实验结果并不一致,分析可能是因为对接过程并没有考虑水和溶剂离子的影响。
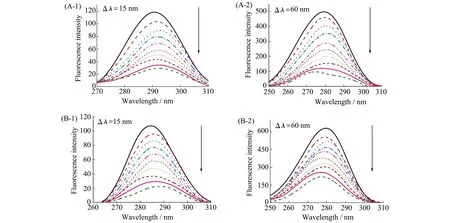
图2 (A)胰蛋白酶和(B)胃蛋白酶在不同浓度I2存在下的同步荧光光谱A-1和B-1为Δλ=15 nm;A-2和B-2为Δλ=60 nm c(胰蛋白酶)=c(胃蛋白酶)= 1.0×10-5 mol/L;c(I2)(从上到下):0、1.0×10-5、2.0×10-5、3.0×10-5、4.0×10-5、5.0×10-5、6.0×10-5、7.0×10-5、8.0×10-5、9.0×10-5 mol/L[31]
因此,使用FL方法研究蛋白质与配体间相互作用主要是根据波长或强度的变化直接或间接地获取信息。同步荧光光谱从最大发射波长变化的角度分析Tyr和Trp残基附近环境的变化,从而推断蛋白质构象变化。结合Stern-Volmer方程,将不同猝灭剂浓度下的荧光强度F和F0代入方程,可以间接计算出结合常数及结合位点数。分子对接可以辅助FL方法,进一步提供配体与蛋白质中氨基酸残基发生作用的类型与距离等数据。
2.1.3CD方法
由于蛋白质等生物大分子具有圆二色性,因此CD光谱在检测蛋白质二级结构变化、分析蛋白质构象变化等方面有着独特的优势[32],可以结合分子对接,从理论模型上进行佐证。已有文献表明,蛋白质的二级结构中,α-螺旋在192 nm处附近有一正谱带,而β-折叠以负谱带的形式出现在216 nm左右,软件拟合后可以求出蛋白质构象中α-螺旋、β-折叠的变化情况[33]。
Zhan等[34]使用CD光谱研究了β-乳球蛋白(β-lg蛋白)结合辣椒素CAP前后二级结构的变化,单独的β-lg蛋白在远-紫外区域的210 nm处有一个负带的吸收,与β-折叠含量有关。加入CAP带来了浓度依赖性的改变,但没有明显的峰位移,表明与CAP的结合没有导致β-折叠的明显改变(图3A)。
Ma等[23]也利用CD光谱分析了蛋白质二级结构的改变,使用圆二色谱数据分析软件DICHROWEB可计算得到。随着SDS的不断加入,α-螺旋的比例增加,而β-折叠减少。与纯的胰蛋白酶溶液对比,α-螺旋比例从9.1%增加到16.7%,而β-折叠含量从32.8%降低到27.6%,考虑到酶活性的降低及二级结构的改变,SDS导致的构象转变很可能发生在胰蛋白酶的活性中心(图3B)。SDS结合胰蛋白酶的对接结果立体图显示(图3C),SDS的疏水烷基链嵌入到靠近活性位点的S1口袋附近,与CD光谱推断的结果一致。
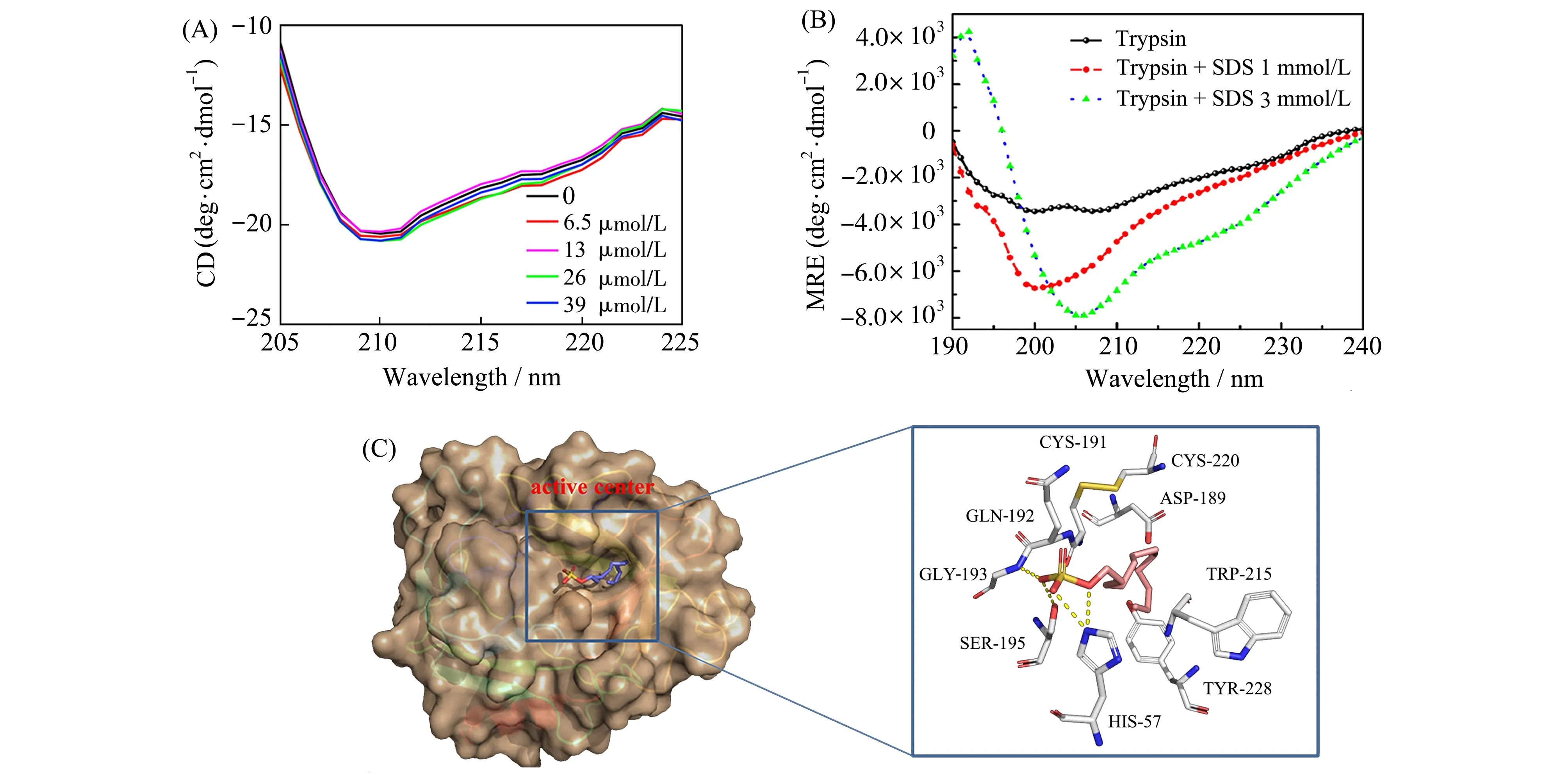
图3 (A)β-lg蛋白在不同浓度CAP存在时的CD光谱[34];(B)胰蛋白酶与SDS的CD光谱[23];(C)SDS与胰蛋白酶的对接结合立体图[23]
2.1.4FT-IR方法
Liu等[26]研究了虾青素ASTA作为配体,分别结合到油酸与BSA复合物OA-BSA和BSA上的情况(图4A、4B)。在ASTA存在时,蛋白质酰胺I带处的吸收峰强降低,归因于BSA的α-螺旋结构减少。OA-BSA和BSA在3429.3、3430.3 cm-1处显示了相似的特征峰,代表O—H的伸缩振动,与ASTA结合后,这部分的峰分别红移至3430.7、3439.4 cm-1,表明ASTA与蛋白质间存在氢键作用。选择最优的分子对接结构进行分析,在ASTA-BSA复合物中,ASTA六元环上的氧原子与Lys294和Arg217上的氢原子形成氢键作用。对于ASTA-OA-BSA的情况,六元环的氧原子与Glu207及Gln33形成氢键作用,验证了FT-IR光谱的结果。该研究利用FT-IR光谱判断了蛋白质二级结构的变化,并通过分子对接说明了蛋白与配体间的氢键相互作用。
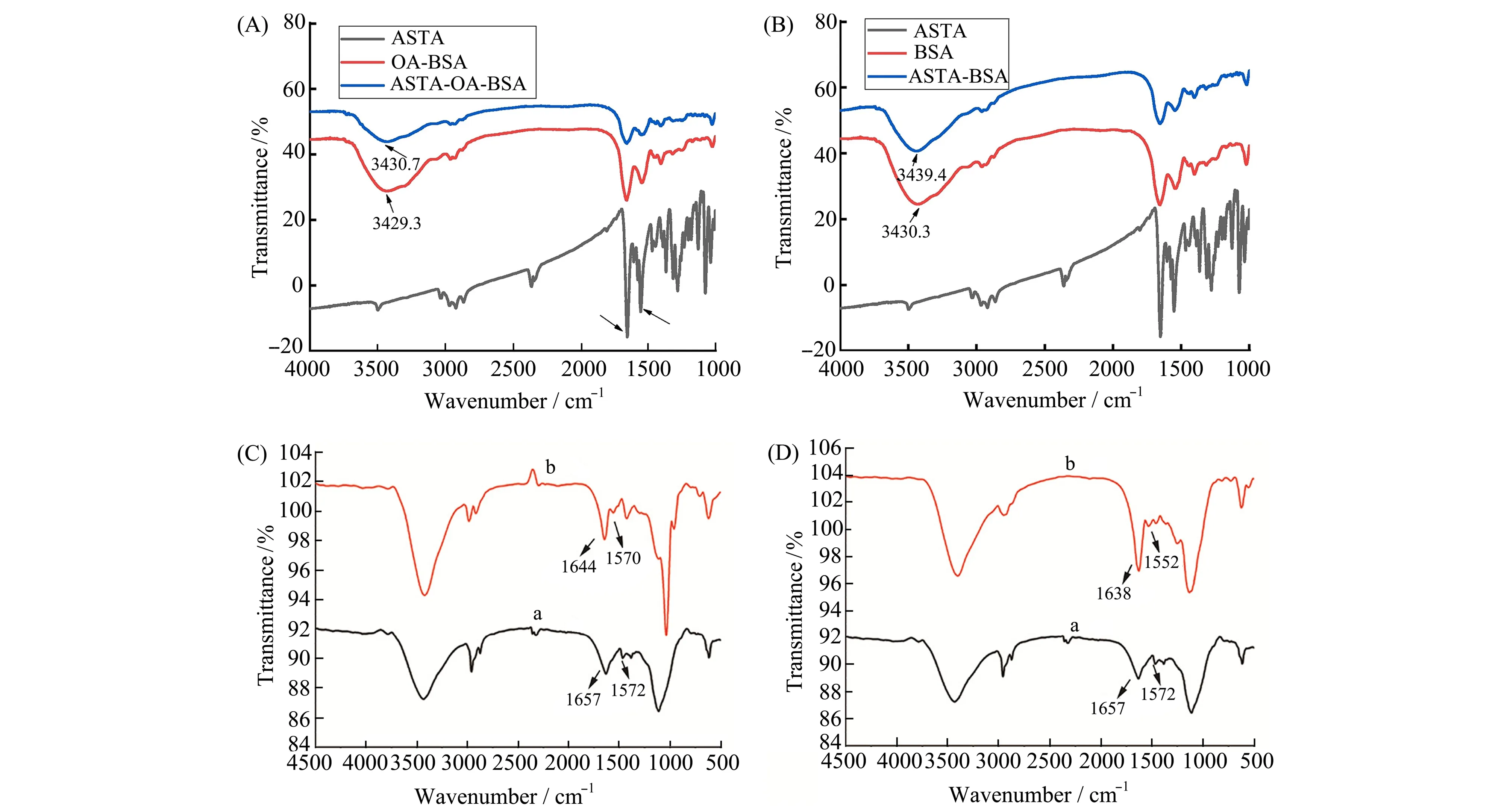
图4 (A)OA-BSA在有无ASTA存在下的FT-IR光谱;(B)BSA在有无ASTA存在下的FT-IR光谱[26];(C)298K下,α-葡萄糖苷酶在(a)没有(b)有圣草次苷存在下的FT-IR光谱;(D)α-葡萄糖苷酶在(a)没有(b)有圣草酚存在下的FT-IR光谱[36]
2.1.53D荧光方法
3D荧光光谱是一种描述荧光强度同时随激发和发射波长变化关系的谱图,同时也是一种表征蛋白质构象变化的可靠科学技术,与UV-Vis光谱、FT-IR光谱、FL光谱,以及CD光谱相比,可以提供更全面的荧光信息[37],对于Tyr和Trp残基尤为敏感。如前所述,使用同步荧光光谱可以得到Trp或Tyr残基附近微环境的变化,CD光谱在分析蛋白质二级结构方面具备独特的优势。而利用3D荧光光谱分析蛋白质与配体相互作用时,可以一次性获取Trp或Tyr残基和多肽主链结构变化的信息。
Han等[38]在研究氟达拉滨Flu与人血清白蛋白HSA作用机制时,使用了3D荧光光谱(图5),分别扫描了游离的HSA与HSA-Flu两个系统,光谱中出现的两个峰被标记为峰1和峰2,峰1反映了HSA的Trp和Tyr残基的固有荧光特征,与三级结构的变化有关。峰2象征着多肽主链结构的荧光特性,与二级结构的变化有关。结果显示,两个荧光峰分别发生了14.6%和45.0%的猝灭,说明Flu与HSA在基态形成复合物,构象发生改变。Flu猝灭HSA的荧光在峰2上比峰1上明显,可以解释为Flu与HSA的相互作用诱导了蛋白质多肽的轻微展开,导致蛋白质的构象发生变化,使蛋白质的疏水区域进一步暴露。分子对接结果显示,Flu的羟基与Val455和Tyr452主链上的氧原子,氨基与Lys212主链的氧原子之间形成氢键。从而验证了3D荧光光谱的结果,一方面Flu与Tyr残基相互作用,另一方面其改变了HSA的微环境并干扰其固有荧光。此外,Huang等[39]使用 3D荧光光谱了解到浓缩乳清蛋白WPC与香豆酸结合时,峰1处的荧光强度下降并伴随有轻微的蓝移,表明微环境更加疏水。与二级结构相关的峰2处荧光强度降低表明两者的结合导致了蛋白质的解折叠,蛋白质的解折叠会使得疏水区域暴露,造成微环境疏水性增加,与峰1处分析的结果一致。而WPC与熊果苷结合时,荧光的变化与香豆酸相反,分析可知Trp氨基酸残基附近亲水性增加,蛋白质骨架发生折叠。
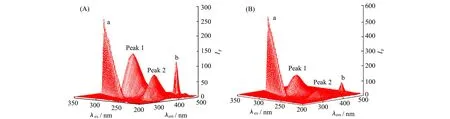
图5 (A)HSA的3D荧光光谱;(B)HSA-Flu系统的3D荧光光谱[38]
2.2 DNA-配体
根据沃森-克里克的碱基配对理论,两条核苷酸链反向平行盘绕,碱基对A-T、G-C互补配对,导致DNA及RNA的螺旋结构中形成有大沟和小沟区域。此外,DNA与小分子的结合模式分为三类:非特异性的静电相互作用;大沟和小沟处的凹槽结合;主要作用力为芳香性分子与碱基对间π-π作用的嵌插结合。DNA与蛋白质遵循相似的理化结合原理,理论上,蛋白质-配体的对接程序应同样适用于DNA与配体的对接。事实上,许多蛋白质包含了定义明确的疏水性结合位点,而核酸拥有更多暴露于溶剂的结合口袋。已有的蛋白质-配体对接程序用于核酸-配体间的对接时,需要进行一定的修改,目前能够用于核酸对接的程序有:Autodock Vina、Gold、Glide、rDock等。
DNA-配体的结合区域或二者间结合模式可以通过多种光谱手段确定,对于具体的相互作用力,可结合分子对接进行模拟计算。脂质-DNA复合物是一种在药物传递中常用的载体。Nandy等[40]结合光谱学与分子对接在分子水平上解释了不同电荷类型的脂质与DNA的相互作用,利用UV-Vis光谱与稳态荧光光谱分析,发现DNA与两类脂质体(正电脂质体、负电与两性脂质体)作用模式不同,DNA与正电脂质体作用更强烈,正电脂质体与小沟处的相互作用更强。为了进一步理解不同类型的脂质体与DNA的不同位点的相互作用,使用PatchDock的对接程序,选择脂质双层与优化的DNA结构进行对接。对接模拟结果推测正电脂质体与DNA的磷酸基团有发生静电相互作用的可能性,以静电作用为主,协同有疏水作用。负电和两性的脂质体与DNA相互作用较弱是因为缺少了静电相互作用(图6A)。
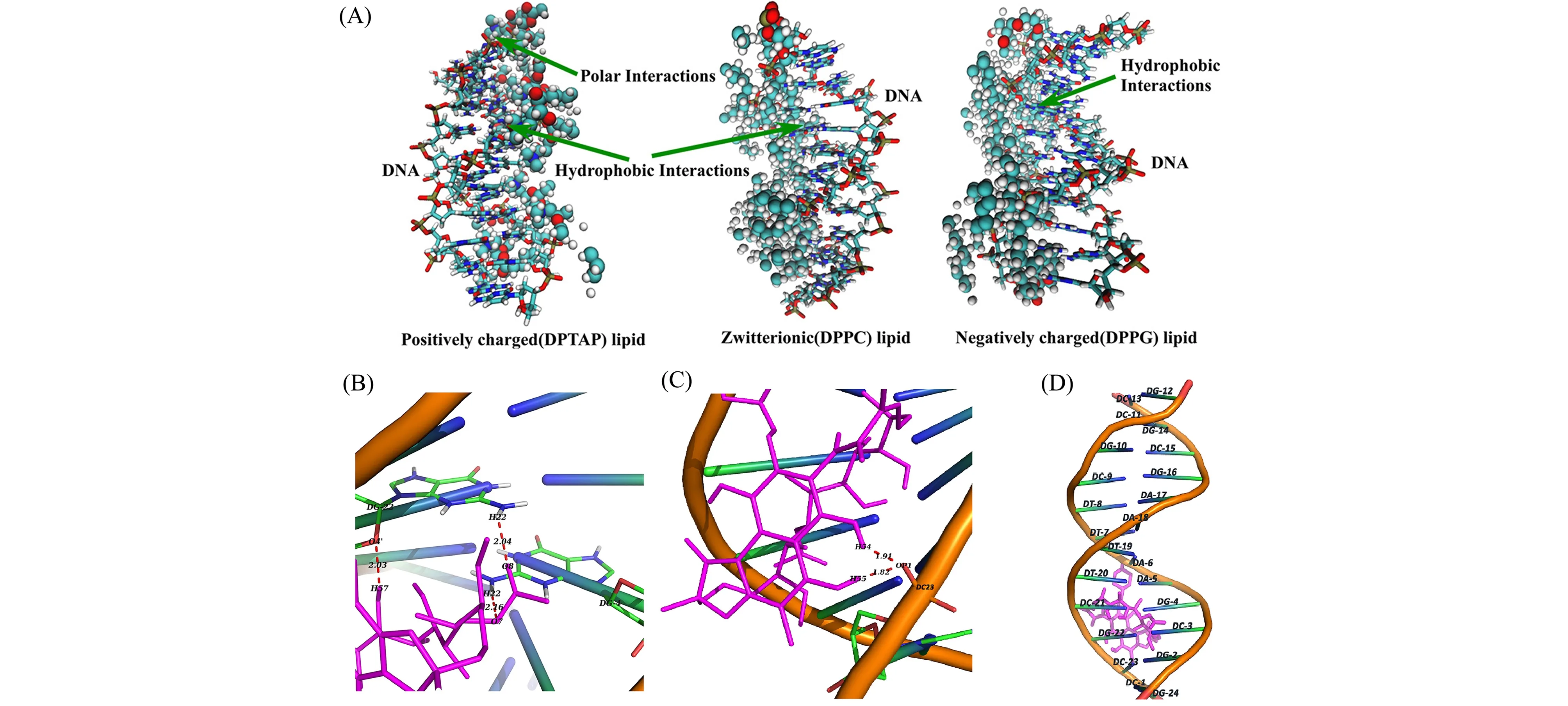
图6 (A)正电脂质体DPTAP、两性脂质体DPPC和负电脂质体DPPG组成的脂质双层与DNA复合物的优化结构[40];(B)和(C)立体观察利福平-DNA对接结构,显示了可能存在的氢键作用;(D)利福平与DNA双链体序列[d(CGCGAATTCGCG)2](PDB ID∶1BNA)间的插入模式表面示意图[43]
此外,对于在小沟和大沟区域均有可能结合的小分子,UV-Vis光谱确定形成复合物后可利用分子对接计算对更有利的结合区域进行评估比较。Javar等[41]报道了一种基于ds-DNA/Eu3+掺杂 NiO/CPE的新型电化学生物传感器,可以用于检测安吖啶。UV-Vis光谱的结果表明安吖啶与DNA有效结合,并通过Benesi-Hildebrand方程[42]计算出结合常数。将安吖啶分别与DNA的小沟和大沟区域进行对接,结果验证了安吖啶在DNA小沟和大沟上均有相互作用,对比结合能结果预测安吖啶更可能结合在大沟区域。
以嵌插作用结合在DNA上的小分子,与DNA的碱基对形成的π-π作用可以改变DNA的CD光谱的特征吸收峰。Chakraborty等[43]研究了药物利福平对CT-DNA的结合模式。CT-DNA的CD光谱在240~400 nm处有两个特征吸收峰,分别是275 nm处碱基π-π作用的吸收,以及245 nm处螺旋带的吸收。如果是小分子以凹槽模式或是以静电作用与CT-DNA作用时,这两处特征吸收峰几乎不会发生变化,因此推测CT-DNA与小分子的结合模式主要为嵌插结合。对接结果分析利福平通过多重氢键与DNA相互作用,并且优先与DNA的富-GC区域结合(图6B、6C、6D)。通常,小分子与DNA的富-AT区域结合表明是凹槽结合模式,与富-GC区域结合是嵌插模式[44,45],综合以上分析,判断利福平以嵌插模式与DNA结合。
2.3 病毒-配体
目前,许多文献报道了分子对接应用于抗病毒药物筛选[46-48],利用分子对接不仅可以进行评分筛选,还可以预测药物的生理和生物活性,以及ADMET(药物的吸收、分配、代谢、排泄和毒性)性质。由于病毒尺寸微小、结构复杂,使用荧光探针对其进行检测具有一定难度,因此,结合光谱学与分子对接用于解释病毒RNA与配体结合机理方面的工作相对较少。
Xie等[49]设计了一种水溶性的二维MOF材料 [Cu(Dcbb)(bipy)(OH)]n(简称MOF1),用于同步检测ZIKV病毒RNA的3′-非编码区域的三个保守靶序列T1、T2、T3。通过公式log[(F0-F)/F]=nlog[Q]+logK计算得到结合常数K,结果表明,MOF1与P-DNA@RNA双链体的结合弱于P-DNAs(ZIKV病毒的全序列)。随后,作者使用Molecular Operating Environment (MOE)软件进行了分子模拟,灵活性更高的P-DNAs能够附着在MOF1的表面(图7A),而P-DNA@RNA双链体由于其自身的螺旋结构与MOF1的接触面较小,显示出“中间隆起和两端接触”的模型(图7B)。P-DNAs和P-DNA@RNA双链体主要通过π-π相互作用与MOF1结合到一起。MOF1与 P-DNAs的π-π相互作用以及氢键作用数量均多于MOF1与P-DNA@RNA双链体。借助UFF力场模拟可知,MOF1与单链P-DNAs (ΔGP-DNAs@MOF) 结合的电子能量远低于与P-DNA@RNA双链体结合的能量(ΔGMOF+P-DNA@RNAs),说明MOF1与单链体结合更紧密,与实验结果一致。
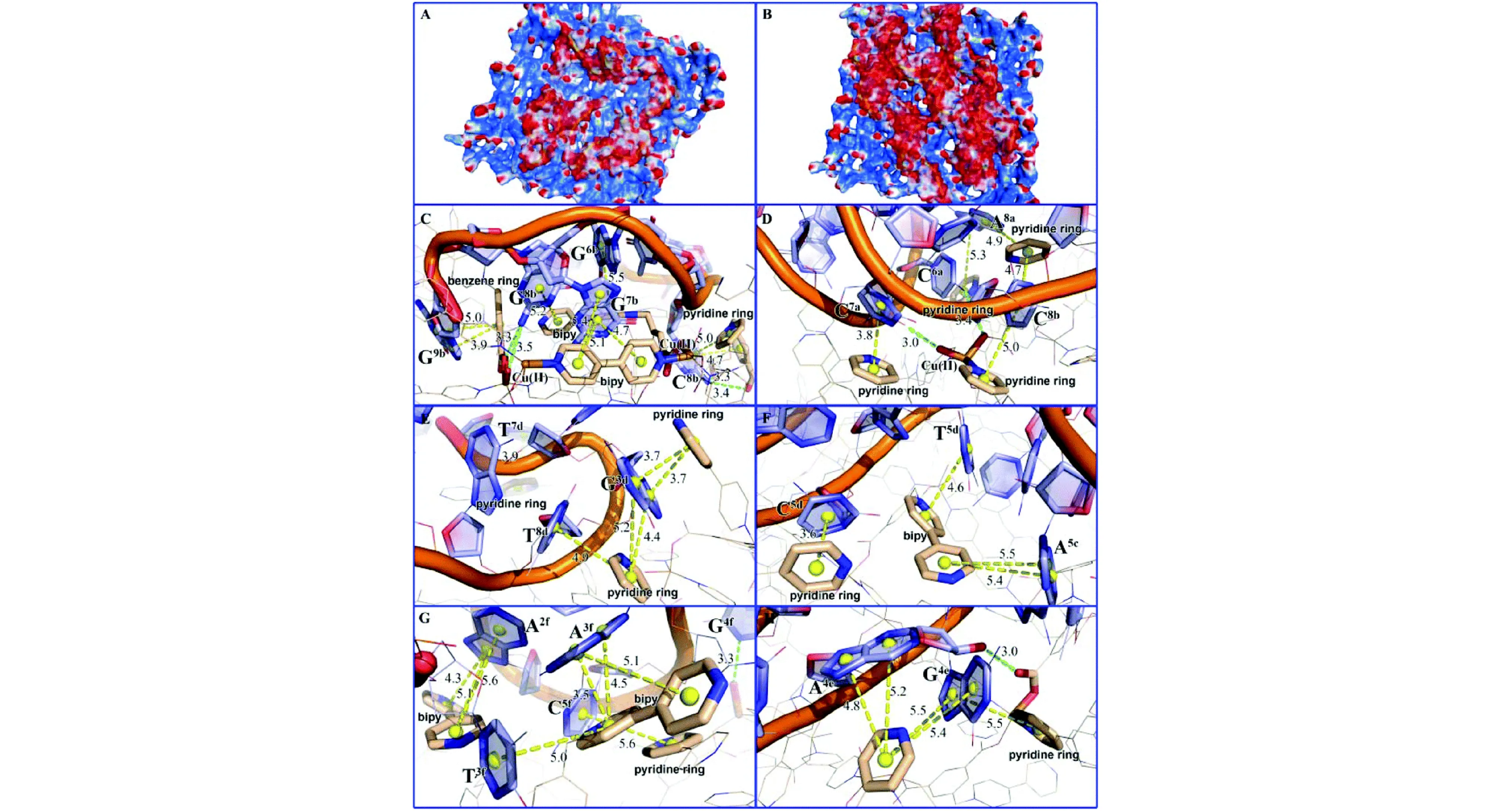
图7 MOF 1与(A)三个P-DNAs(B)三个P-DNA@RNA双链体间的相互作用,红色、蓝色和白色区域分别代表负电、正电和中性表面;(C)、(E)和(G)分别代表MOF 1与三个P-DNAs的结合模式;(D)、(F)和(H)分别代表MOF 1与三个P-DNA@RNA双链体的结合模式[49]
3 总结与展望
综上所述,通过光谱学的分析技术和分子对接软件相结合能够很好地解释蛋白质及核酸等生物大分子与配体间的相互作用,为小分子探针的分子结构设计奠定了基础。对于蛋白质与小分子配体间相互作用的研究工作进行的较多,能够推导出配体对蛋白质二级结构的影响(包括α-螺旋结构、β-折叠等),分析发色团Tyr和Trp残基附近微环境的变化或是结合热力学方程计算得到结合常数等热力学性质。对于DNA或病毒,相应的研究工作还有待于进一步发展,目前可以获取配体结合的区域、结合模式或结合作用强弱等信息。
分子对接作为一种传统的药物设计方面的计算软件,与光谱学手段相结合,在蛋白质、DNA、病毒RNA与小分子结合的机理解释方面已经被越来越多的人使用,可以弥补光学方法不够直观的缺点,直观地呈现出结合位点、结合作用力类型及相互作用距离等,这种学科间的交叉结合,彼此间的互补可以从多角度阐述问题。分子对接计算的是受体与配体在真空中的结合情况,但对于一些更加复杂的体系和环境,尤其是溶剂对结合体系影响较大时,分子对接得出的结果准确度仍有待提高,仍需要不断去优化算法参数等,必要时结合分子动力学模拟等手段进行辅助分析。
