美国医疗器械管理类别动态调整机制及其工作思路探究
2021-06-11张春青周良彬王越田利余新华计雄飞张辉
【作 者】张春青,周良彬,王越,田利,余新华,计雄飞,张辉
1 中国食品药品检定研究院,北京市,100050
2 广东省医疗器械质量监督检验所,广州市,510663
3 国家市场监督管理总局国家标准技术审评中心,北京市,100088
0 引言
2018年8月1日,我国新修订的《医疗医疗器械分类目录》正式实施,新分类目录按照《医疗器械分类规则》制定完成,在一定时期内《医疗器械分类规则》是相对稳定的,为保证医疗器械监管的一致性和连续性提供了技术保障。但是,随着医疗器械行业的快速发展,新技术、新产品的不断涌现,在医疗器械产品的生产、经营、使用过程当中,生产企业和监管部门对产品的认识也不断积累经验,基于“新信息”,会对产品风险进行重新评估,根据产品风险程度的判断,对部分产品管理类别进行动态调整,以适应日新月异的行业发展带来的监管新需求和挑战,实现医疗器械分类的精准性和科学性。
美国作为医疗器械监管的先行者,其管理类别动态调整机制[1]从1976年建立至今,通过动态调整维持了其医疗器械分类系统的稳定有效运行。研究旨在通过回顾2013—2018年6年间,美国FDA对部分医疗器械风险进行重新论证,实施动态调整的工作思路和具体流程,为我国相关工作提供借鉴和参考。
1 工作基础和依据
2012年7月9日,美国国会颁布了“食品药品监督管理局安全与创新法案”(Food and Drug Administration Safety and Innovation Act,FDASIA),对FD&C法案进行修订[2]。FDASIA第608(a)节修订了FD&C法案第513(e)节,将医疗器械管理类别调整程序的性质由规则制定变更为行政命令,此前管理类别调整文件为规则(Rule),之后则为指令(Order)。FD&C法案第513(e)(1)(A)(i)节随后据此更新,规定了发布正式指令的程序,具体来说,在发布行政指令将医疗器械管理类别调整之前,须进行以下操作:①在联邦公报(Federal Register)中公布管理类别调整的指令草案;②医疗器械分类专业组召开会议;③综合考虑公众评论。医疗器械管理类别调整的指令草案必须列出拟定调整的管理类别,以及管理类别调整有效科学证据(valid scientific evidence)的概要,包括使用医疗器械的公共卫生利益,以及该医疗器械风险的性质和发生概率[1]。为了适应这一法规变化,FDA修订了其分类程序文件21CFR第860部分[3],纳入以上FDASIA最新要求,更新了管理类别调整程序,新增了由FDA主动发起的管理类别调整程序,其管理类别调整程序不再限于对相关请求的回应。
作为管理类别调整依据的FD&C法案第513(e)(1)(A)(i)条规定,FDA可以通过行政命令,根据“新信息”对医疗器械的管理类别进行调整。其中所指“新信息”一词包括对原有分类数据重新评估而产生的信息,以及医疗器械原先分类时所没有的信息。重新评估之前的数据是后续监管行动的基础,这样的重新评估是基于新的监管思路或医疗科学的变化。无论数据新旧,根据FD&C法第513(e)节,用于支持管理管理类别调整的“新信息”必须是“有效科学证据”。如FD&C法案第513(a)(3)节的和21 CFR第860.7(c)(2)节所定义:有效科学证据来自良好对照研究、部分对照研究、无匹配对照的客观试验及研究、由合格专家进行的详细记录的案例历史以及已上市医疗器械的重要使用经验报告,合格的专家可以据此公平合理地确定医疗器械在其使用条件下的安全性和有效性。所需证据可能由于器械特性、使用条件、警告和其他限制的存在及充分性、使用经验而有所不同。孤立的案例报告、随机经验、缺乏细节而无法进行科学评估的报告以及未经证实的意见,不能被视为体现安全性或有效性的有效科学证据。然而,在识别安全性和有效性值得怀疑的器械时,可以参考这样的信息。在分类过程中,FDA依靠“有效科学证据”来确定医疗器械监管水平。在管理类别调整过程中考虑的、FDA所依据的“有效科学证据”必须公开提供。公开信息不包括商业机密信息,例如,待批准的上市前审批申请相关内容。
2 工作文件要素
根据“有效科学证据”确定调整产品类别之后,相关决定形成管理类别调整文件面向公众征求意见后正式发布。不管是作为规则还是指令,管理类别调整文件通常包括以下内容:法规背景、器械信息、管理类别调整影响分析、拟修改的分类条款正文、1995文书削减法案相关内容、参考资料。随着监管思路的完善,以及信息公开的需要,近年的管理类别调整文件中,对分类理由、讨论过程、公众意见及处理等方面信息的阐述更为详尽,部分文件根据需要对相关产品的风险识别以及风险控制措施进行了具体的阐述(风险识别和控制措施通常出现在降类文件中,如:体外心脏挤压器),文件信息丰富程度及篇幅普遍大幅度增加。
面向公众征求意见的指令草案侧重于对调整管理类别的理由进行说明(主要为风险点的论证,如经阴道放置的盆腔器官脱垂用妇科泌尿外科网的分类指令草案中对其风险的详尽讨论),而最终规则/指令则更多篇幅用在说明对所征集意见的处理和解释,以及产品风险水平的界定和所需管控措施的确认等方面。
3 近年工作成果概况
本小节汇总分析了作为监管方的FDA对高风险产品的整体回顾性审核,经评估后降类产品的落实情况,2013—2018年6年间完成管理类别调整的产品情况。
3.1 2014—2015年对高风险产品的整体回顾性审核
为取得行业发展和市场监管的最佳平衡,促进和加快高风险医疗器械发展和审批,2014—2015年,FDA对当时已上市的、需要上市前审批(PMA)的Ⅲ类高风险医疗器械进行了全面的回顾性审核评估,基于这些产品临床研究、非临床数据以及实际临床表现等信息,评估上市前/上市后数据要求对于其安全性的影响水平,并针对不同情况提出了相应的调整方案[4-5],其中明确拟由Ⅲ类调整为Ⅱ类的产品有34个[6],目前仅完成5个(具体见表1),完成率为14.7%。
3.2 2013—2018年管理类别调整情况汇总
FDASIA第608(c)节要求FDA每年公布上年度管理类别调整的器械数量和类型。为符合这一法规要求,FDA官方网站以列表形式给出了从2013年(即FDASIA颁布后的第一个完整年度)开始至今完成管理类别调整的所有器械列表[7]。FDA将在每次管理类别调整完成后更新表格。基于以管理类别调整列表中链接的管理类别调整指令,查询联邦公报网站[8]以及FDA产品分类数据库[9],对产品管理类别调整情况及调整类别经历的时间等相关信息进行了汇总,具体内容见表2。
如表2所示,2013—2018年,FDA通过发布指令,对25个产品进行了管理类别调整,降类20个,升类5个。截至2019年7月,FDA产品分类数据库产品总数为6 530,2013—2018年的6年时间调整管理类别产品占比约为0.38%。按照表2的数据统计,从发布拟调整管理类别的指令草案征集公众意见,到正式发布调整指令,所用时间从6个月到40个月不等,平均为20.5个月(产品升类平均为23.4个月,产品降类平均为19.8个月)。对各产品管理类别调整用时统计表明,不管是升类还是降类,都是一个耗时较长的过程。
4 案例分析
4.1 降类举例
随着行业对某一类产品了解的加深,收集到该类产品安全性和有效性相关的数据不断丰富,FDA从之前对产品逐个评估其风险-收益特征,逐渐总结归纳该类产品共同风险点。在以上工作基础上,如果产品主要风险可以通过制定并执行该类产品通用的特殊控制措施(必要时,结合一般控制措施)而得以控制,建立合理的安全性和有效性保证,该类产品经过评估后,可从Ⅲ类降到Ⅱ类。如果Ⅱ类产品被认为执行一般控制措施即可建立其安全性和有效性的合理保证,则其管理类别可能被调整为Ⅰ类。本小节以体外心脏挤压器(DRM)为例,根据产品预期用途,对其风险进行识别和有效控制措施,全面阐释了管理类别降低的研究论证过程和考虑因素。
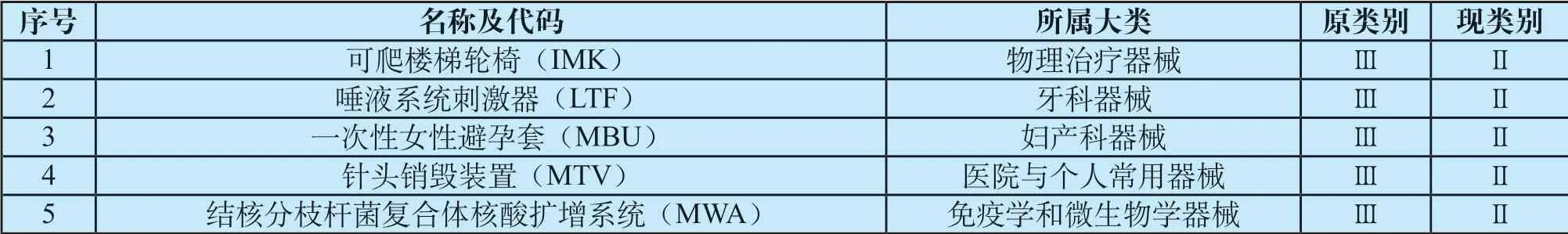
表1 2014—2015 年 FDA 对高风险医疗器械回顾性审核中已经完成降类的产品 Tab.1 Reclassified products in FDA's retrospective review of high-risk medical devices
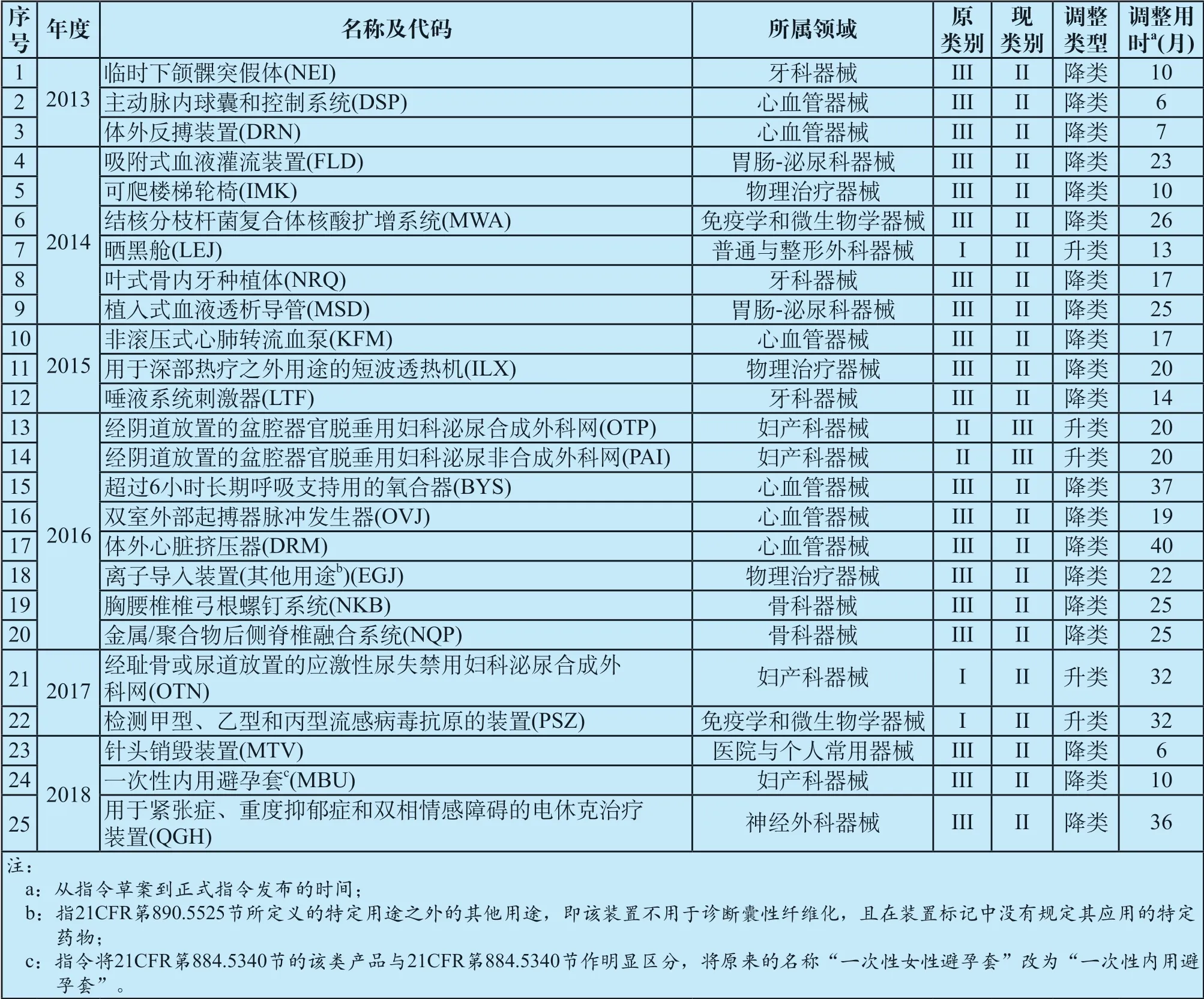
表2 2013-2018年FDA医疗器械管理类别调整情况统计 Tab.2 Summaries of FDA medical device classification adjustments from 2013 to 2018
在分类程序文件21CFR中,关于体外心脏挤压器的描述是:施用于患者外部的处方器械,可以为电动、气动或手动,用于在患者心脏区域间歇性地压缩胸部以在心脏停搏期间提供血液流动。当有效的手动心肺复苏术(cardiopulmonary resuscitation,CPR)不可能实施时(例如当患者在转运途中需要持续的CPR,或可以提供有效CPR的医疗人员不足时),体外心脏挤压器作为手动心肺复苏的补充使用。
体外心脏挤压器管理类别调整指令草案[10]中,FDA提出了该类产品的风险主要存在于组织/器官损伤、血流不足、心律失常或电击、皮肤不良反应四个方面,且现在已有足够的有效信息来建立特殊控制措施,包括设定医疗器械的特定使用人群、标明使用条件、限定由经过适当培训人员使用,以及确保医疗器械的完整性能测试等方面的控制措施,从而确保得到足够的胸部压缩频率和深度,充分降低该类器械的风险。FDA认为这样的特殊控制措施,结合一般控制措施,足以提供合理的安全性和有效性保证。因此,根据FD&C法案的第513(e)和515(i)节和21 CFR 第860.130节,基于医疗器械的相关新信息,FDA主动提出将该器械管理类别由Ⅲ类降为Ⅱ类。
正式指令[11]中,以主要篇幅阐述了FDA和专业组对公众意见的回复、产品风险的识别以及特殊控制措施的确定。在面向公众征集意见的基础上,经FDA与体循器械专业组的反复讨论,并对指令相关内容不断修改,最终确认该产品的风险已充分识别,制定了针对性的风险控制措施,并作为该产品的特殊控制要求写入正式指令(体外心脏挤压器的风险及控制具体内容见表3)。在FDA与专业组就风险识别和控制措施内容取得一致认识,且对公众意见能作出适当回应与解释的情况下,FDA认为产品执行一般控制和特殊控制措施可以提供产品安全性和有效性的合理保证,最终确定将该类产品管理类别从Ⅲ类降为Ⅱ类,并发布了该类产品的特殊控制要求。
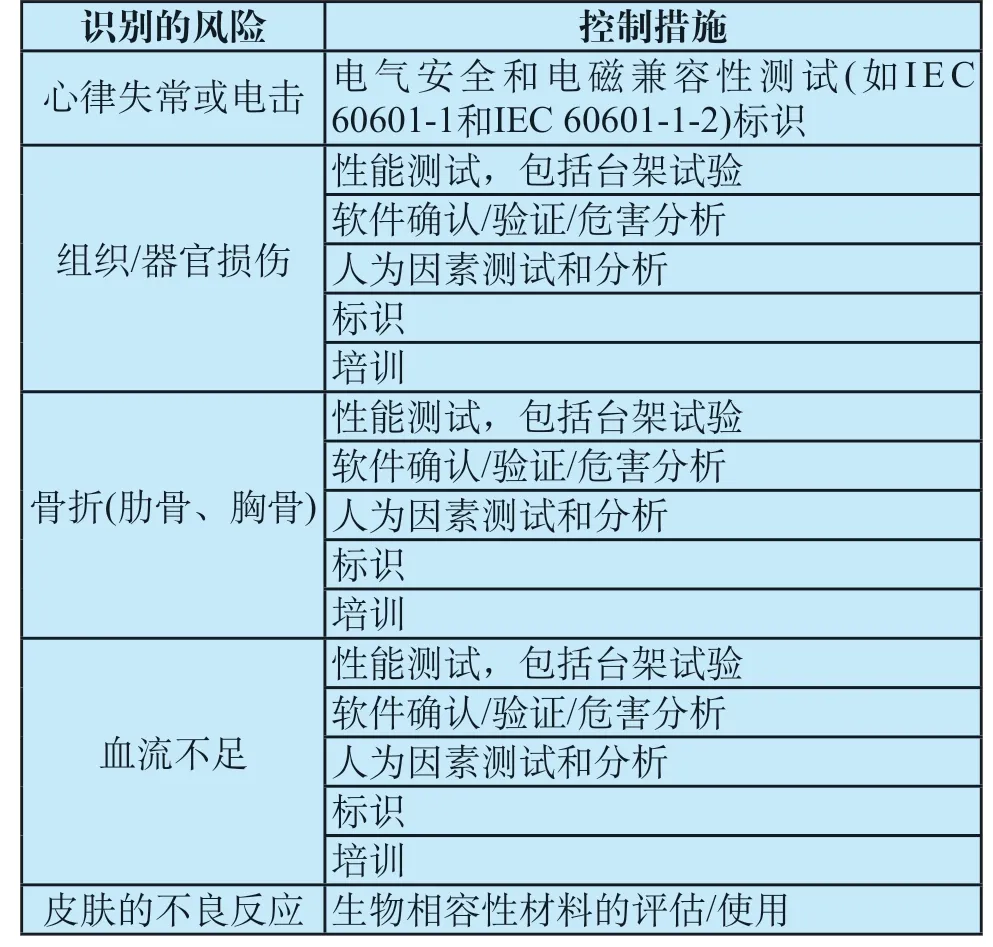
表3 体外心脏挤压器类医疗器械风险及控制措施Tab.3 Risks to health and mitigation measures for external cardiac compressor devices
4.2 升类举例
如果作为“有效科学证据”的新信息显示,产品现有管控措施不能建立起针对其预期用途的安全性和有效性的合理保证,FDA或相应的专业组会建议启动产品管理类别调高的工作程序。本小节详细阐释经阴道放置的盆腔器官脱垂用妇科泌尿外科网(合成OTP/非合成PAI)产品升类论证分析、征集意见和发布正式调整管理类别指令的过程。
在分类程序文件21CFR中,关于盆腔器官脱垂用妇科泌尿外科网(合成OTP/非合成PAI)的描述是:用于加强骨盆底部软组织的处方器械。该器械为合成的、非合成的或两者兼有的多孔植入物。该器械不包括用于其他预期用途的外科网。
该产品管理类别调整指令草案[12]中,以主要篇幅引用大量临床研究和不良事件数据来作为讨论产品安全性和有效性的“有效科学证据”。第一,安全性方面。FDA通过对产品并发症报道、随机对照试验等公开文献以及不良事件数据等信息的整理分析,明确该类产品存在潜在不合理的疾病或伤害风险,主要在三个方面存在风险,分别是:①术中风险(穿孔、出血);②手术网在阴道中暴露(在阴道上皮可见手术网,可造成骨盆疼痛、感染、阴道再次出血等,往往需要二次手术);③手术网在阴道中被挤压(手术网进入膀胱、直肠等内脏器官,造成骨盆疼痛、感染、瘘管形成等,往往需要二次手术)。第二,有效性方面。FDA对该类产品有效性的资料进行分析后,认为依据现有临床资料,不能给出该类产品具有比传统的、非手术网修复方式更有效的结论。基于以上分析,FDA和专业组一致认为这一产品类型的安全性和有效性尚未确定,正面的风险-收益特征(benefit-risk profile)未能建立(即不能确定该类产品带来的收益高于风险)。根据FD&C法案第513(a)(1)(C)节和21CFR第860.3(c)节,FDA认为该类产品现有的信息不足以确定一般控制和特殊控制能够提供安全性和有效性的合理保证,并且考虑其用途对于人生命健康具有重要意义,又具有潜在不合理的疾病或伤害风险,符合法规关于Ⅲ类产品的表述,应将其管理类别调整为Ⅲ类。每个器械应在临床研究中进行评估,通过与原生组织修复方式进行比较分析,建立其安全性和有效性合理保证,并在相关资料中同提供研究获得的产品风险-收益特征相关信息。
正式指令[13]中,主要介绍公众对指令草案的反馈意见和FDA在对各方面意见疑问进行解答的基础上,确认了一般控制和特殊控制不足以为该类器械提供合理的安全性和有效性保证,而且在产品未建立积极的利益-风险概况的情况下,该类产品的使用表现出潜在不合理的疾病或伤害风险。正式指令最终确定该产品管理类别由Ⅱ类调整为Ⅲ类,并发布了最终的分类规章及PMA要求。
5 总结与展望
按照前文数据的统计分析,美国FDA虽然多年持续执行了医疗器械管理类别动态调整机制,但是实际调整管理类别产品的数量较少,表明FDA医疗器械分类系统具有整体稳定性,监管方和行业相关方都注重保持产品监管要求的长期一致性。同时,对各产品管理类别调整用时统计表明,不管是升类还是降类,都是一个较为耗时的过程。说明管理类别作为对产品监管水平的基本定位,想要对其做出调整,需要在充分收集相关信息、行业各方面充分论证的前提下才能最终得以确认。调整管理类别的工作,不仅要响应行业对产品日益积累不断加深的认识,对安全性和有效性进行及时的风险评估,也要综合考虑公共卫生利益和企业合规成本之间更为合理的平衡。最后落实到实际中能够调整管理类别的产品,都经历了一个长期的慎重的论证过程。
美国FDA的医疗器械管理类别动态调整机制已经实践多年,其中专业组和FDA的互动关系和管理类别调整工作程序、分类调整草案和正式文稿的起草与修改、“有效科学证据”的分析确立、管理类别调整论证思路和逻辑等相关内容,在一定程度上可以作为我国医疗器械管理类别动态调整机制建立和完善过程中的参考和借鉴,以建立和完善更加科学的医疗监管体系,使得监管政策和行业产品发展之间建立更加良性的互动关系,不断促进医疗器械领域的创新和发展,也为优化监管资源配置提供依据,最终为满足不断发展的临床使用需求、为实现健康中国发挥更好的作用。
美国医疗器械管理类别动态调整工作思路是在最新的有效科学证据基础上,以风险识别和控制为依据,以提供更加合理、有效的产品安全性和有效性保证为目的,行业相关方展开反复讨论并取得最终一致。该工作机制经多年实践,已相对成熟,其程序和工作思路可以作为我国工作参考和借鉴。
