先天性与获得性长QT综合征诊断治疗现状
2021-06-08李翠兰刘文玲高元丰
李翠兰 刘文玲 高元丰
(1.北京大学人民医院心内科,北京 100044; 2.首都医科大学附属北京朝阳医院心脏中心,北京 100020)
长QT综合征(long QT syndrome,LQTS)是一种罕见的心脏离子通道病,表现为心电图上QT间期延长,T波异常,易产生恶性心律失常[如尖端扭转型室性心动过速(torsade de pointes,TdP)和心室颤动等]、晕厥甚至心源性猝死的一组综合征[1]。根据病因不同可分为先天性LQTS(congenital LQTS,cLQTS)和获得性LQTS(acquired LQTS,aLQTS)两大类。cLQTS是引起青少年猝死的重要原因,目前临床主要通过心电图特征表现和临床特点,并结合基因检测结果进行诊断及亚型鉴别。比cLQTS更常见的是aLQTS,多继发于电解质紊乱和使用延长QT间期的药物等。现针对cLQTS各方面的最新进展进行综述,之后讨论在当前全球新型冠状病毒流行期间新型冠状病毒肺炎(corona virus disease 2019,COVID-19)患者发生心律失常的潜在危险因素,以及为避免引起aLQTS,在对其进行治疗时可采取的风险评估流程。
1 cLQTS的诊断标准
cLQTS患者的诊断依据包括家族史、不明原因晕厥、心电图上QTc延长以及基因筛查结果。首先了解患者是否有晕厥、癫痫或心源性猝死的病史,以及直系亲属中是否有这些病史。对于QTc处于临界值的患者,需进一步做运动试验及动态心电图检查以及基因筛查。根据2015年ESC发布的室性心律失常和猝死预防指南[2],LQTS的诊断标准包括:(1)在12导联心电图上证实患者QTc≥480 ms,或LQTS风险评分≥3.5分(表1)[3-5];(2)发现明确的LQTS相关致病基因突变;(3)除外继发因素,多次重复12导联心电图提示QTc≥460 ms,并伴有不明原因晕厥。

表1 cLQTS诊断标准:Schwartz评分量表2012更新版
2 cLQTS的基因分型及临床表现
cLQTS按照是否伴耳聋又可分为两种形式:Romano-Ward综合征(RWS)和Jervell-Lange-Nielsen综合征(JLNS)。RWS最为常见,为常染色体显性遗传,后代患病概率为50%。心电图上表现为QT间期延长,T波电交替,发作时出现TdP,临床表现为晕厥和猝死等。多数仅有心脏方面的异常,少数亚型可伴有非心脏异常。JLNS发病率相对较低,为常染色体隐性遗传,即父母双方各带一个相同或不同的LQTS相关突变,然后同时把突变传给子代,这种情况下子代的患病概率理论值为25%。临床上除与RWS一样的心脏表现外,还有神经性耳聋。由于患者携带两个突变的累加效应,通常这种亚型的患者临床症状更严重,发生致命性心脏事件的概率也更高[6]。
目前至少已确定了15个与RWS相关、2个与JLNS相关的基因,与药物引起LQTS相关的基因也有报道(表2)。基因筛查的阳性率约75%,其中LQT1、LQT2和LQT3是最常见的LQTS亚型,占到全部已知基因型病例的90%以上。
3 cLQTS各主要亚型的特征性临床表现及危险分层
研究发现cLQTS各主要基因型多有特征性临床表现,概括而言,LQT1患者心电图多为T波基底部宽大,交感神经兴奋时发病;LQT2患者则表现为心电图上双峰T波,情绪刺激发病为主;LQT3患者主要是心电图上ST段延长及不对称高尖T波,多为静息时发病。以下详细介绍各亚型特点。
3.1 LQT1
LQT1患者更容易在交感神经兴奋时(如游泳和潜水等运动或情绪紧张)发生心脏事件。正常人交感神经兴奋可激活IKs通道,从而加快心室复极过程,QT间期随之缩短。但LQT1患者携带的KCNQ1基因突变,导致IKs缺陷,因而心室复极或QT间期不能随心率增加而缩短,引起QT间期延长。LQT1患者心电图可表现为T波宽基底、高尖和不对称的特点。其ST-T改变可有四种模式:(1)婴儿型:ST-T段短促,与T波上升支融合,呈直斜线状,T波基部较宽,顶部尖锐,T波下降支陡立,呈非对称状;(2)宽大T波:T波呈单峰,基部宽大,上升支和下降支光滑;(3)正常T波:T波形态表现正常;(4)晚发正常T波:ST段延长,T波形态正常,QT间期明显延长[7]。儿茶酚胺诱发试验或运动平板试验可识别LQT1亚型,平板运动试验时,LQT1患者的QTc在恢复期(2~4 min)可出现进一步延长,低剂量肾上腺素注射[<0.1 μg/(kg·min)]可使QT间期的绝对值延长>30 ms[8]。
3.2 LQT2
LQT2患者往往在情绪激动(49%)或突然出现听觉刺激(如铃声和打雷等)(49%)后出现室性心律失常,睡眠中(22%)和运动(29%)诱发症状相对少见。女性在经期和产后特别容易出现心律失常[9]。LQT2的主要心电图特征是多导联双峰T波,T波幅度常偏低,QT间期可为正常或明显延长。双峰T波可分为四种形态:(1)明显型双峰T波:T波两峰分明,第2峰常位于T波下降支的早期(I型)。(2)表浅型双峰T波:T波双峰(或切迹)表浅,有两种形态,第2峰可位于T波顶部(Ⅱ型)或下降支上(Ⅲ型)。(3)低钾血症型双峰T波:T波低平,两峰间距离较大,第2峰常与U波融合,类似于低钾血症时的心电图改变(Ⅳ型)[7]。
3.3 LQT3
LQT3患者多数(约65%)心律失常事件发生在睡眠或休息时,心率越慢QTc越长,越容易诱发心律失常事件。LQT3的心电图有两种ST-T改变模式:(1)晚发尖锐/双向T波:ST段平直或斜性延长,T波尖锐,起始和终止分明,双向T波常见,QT间期多为显著延长。(2)非对称高尖T波:T波高尖,下降支陡立,呈非对称型,QT间期正常或明显延长[7]。
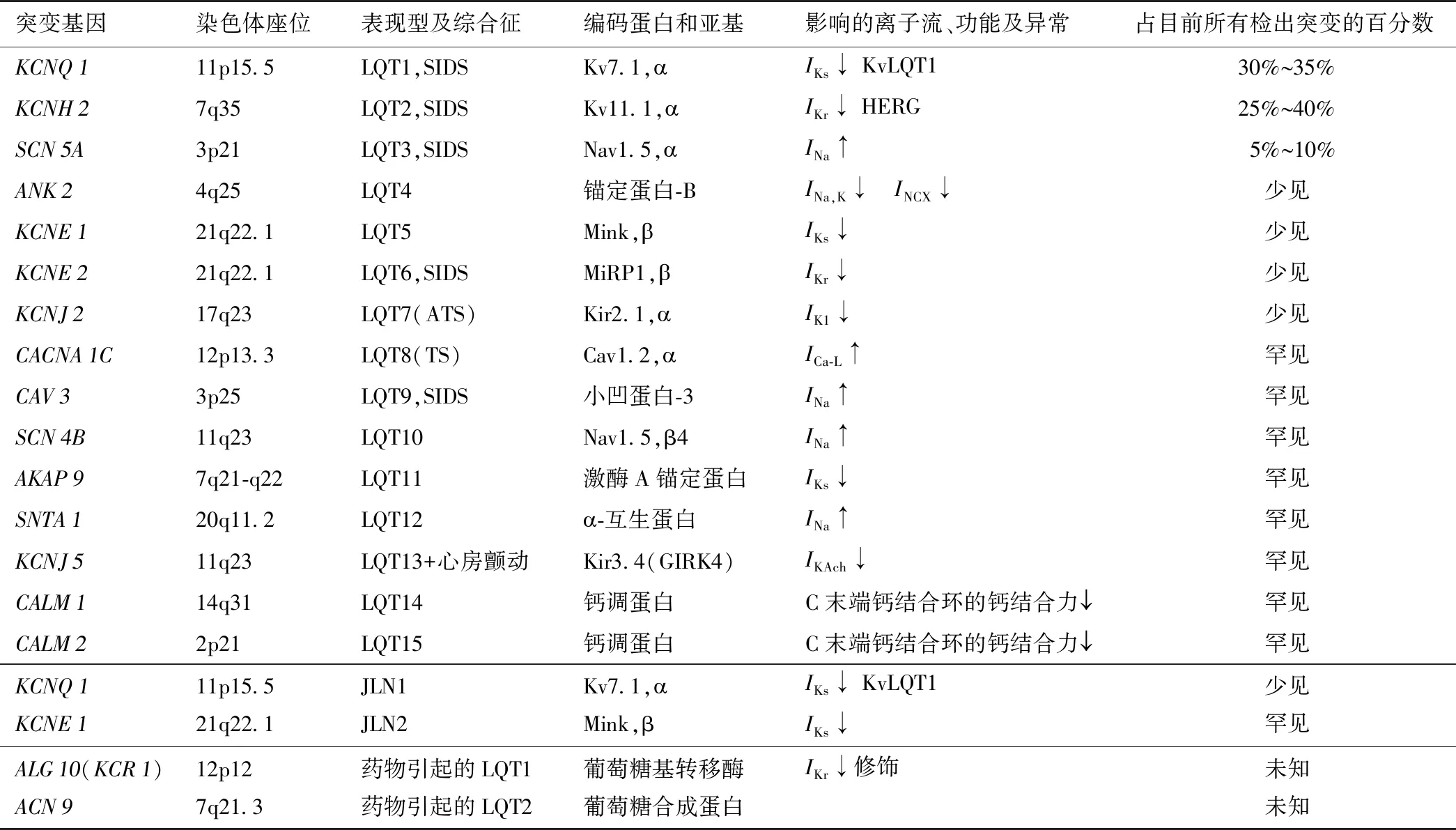
表2 LQTS的基因分型
3.4 其他典型LQTS亚型
LQT7,即Andersen-Tawil综合征,临床特点为QT间期延长、周期性麻痹和骨发育不良。骨发育不良包括矮小身材、脊柱侧突、指(趾)弯曲、眼距过宽、小或大耳伴耳位低下或倾斜、小颌和宽额。有些患者还有心脏本身结构病变,如二叶式主动脉瓣伴或不伴主动脉缩窄或肺动脉瓣狭窄[6]。
LQT8,又称Timothy综合征,表现为多器官异常。心脏异常包括QT间期延长、室性心动过速、窦性心动过缓、房室传导阻滞、动脉导管未闭、卵圆孔未闭、室间隔缺损、法洛四联症和心脏肥大等。中枢神经系统异常包括孤独症、智力发育迟缓和癫痫。脸部异常包括圆脸、低鼻梁、上颚退缩和上唇薄。表皮异常:并指/趾及无毛等。此外还会伴有低血钙、低血糖、低体温和肌无力等,其中QT延长和并指/趾是Timothy综合征1型必有体征[6]。后来也有报道同样基因上的不同突变不伴并指/趾的病例[10]。
另外,KCNQ1和KCNE1纯合或复合杂合基因突变所引起的JLNS,亦有典型的心律失常外症状,主要表现为严重的感音神经性耳聋。
3.5 cLQTS的性别差异
据估计cLQTS患病率约为1∶2 000[11],女性患者稍多于男性(1.6~2.0∶1)[12]。有关对后代的影响,Cuneo等[13]发现,母亲患有LQTS时发生胎儿死亡的概率明显高于父亲为LQTS患者的情况(24.4% vs 3.4%,P=0.036)。LQTS孕妇怀孕期间发生死产(>20周的胎儿死亡)的概率是正常孕妇的8倍(4.0% vs 0.5%),而流产(<20周的胎儿死亡)的概率是正常孕妇的2倍(16% vs 8%)。该研究首次提供了胎儿死亡的病因学证据,提示LQTS引起的离子通道异常可能会导致胎盘和子宫肌层的功能异常。
3.6 cLQTS危险分层
Shimizu等[14]于2018年报道,在1 124例有致命性心律失常的日本LQTS患者中,发现基因型特异风险存在性别差异;年龄<15岁时无差异,15岁之后LQT1/LQT2患者都是女性风险更高。对于LQT1患者,携带位于KCNQ1通道跨膜孔区的致病变异比位于C-末端的变异有更高的发生心律失常的风险(HR1.60,95%CI1.19~2.17,P=0.002),尽管这个现象仅在女性患者身上观察到。对LQT2患者,携带位于KCNH2通道S5-孔区-S6区域突变的患者发生心律失常的风险比携带其他区域的突变要高(HR1.88,95%CI1.44~2.44,P<0.001),且无性别差异。青春期之后的LQT2女性患者风险明显增加(55.2% vs 20.2%,P<0.001)。对LQT3患者,位于Nav1.5通道上S5-孔区-S6区域的致病变异致心律失常的风险比其他位点的变异高(HR4.2,95%CI2.09~8.36,P<0.001),亦无性别差异。
LQT1~3亚型患者在遗传诊断、发生TdP的风险、治疗策略及预后等方面都存在基因型特异的差别。总的来讲,青春期之后女性的QTc比男性长。当QTc<500 ms时,年龄<13岁的男性、年龄>13岁的女性LQT1患者及所有女性LQT2患者都有中等风险;QTc≥500 ms时,不管哪种基因型都是高风险,尤以携带位于通道跨膜孔区及附近的错义突变时风险最大(见图1)[15]。尚无有关LQT4~15亚型患者的风险评估数据。
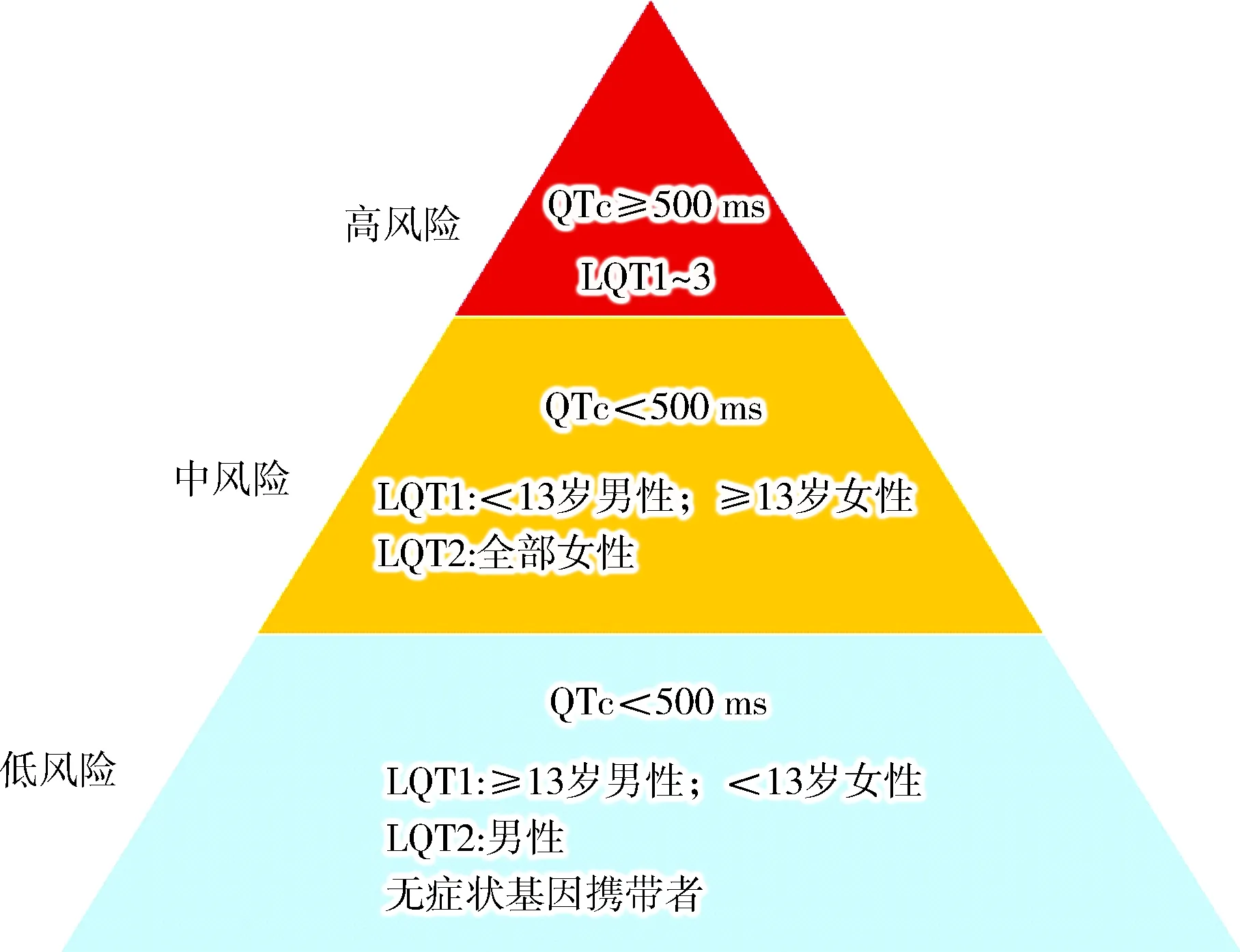
图1 基于基因型、年龄和性别的cLQTS危险分层策略注:QTc≥500 ms的患者携带位于通道跨膜孔区及附近的错义突变时风险最大。
4 cLQTS的治疗
所有LQTS患者,无论是否有症状,都应首先注意改变生活方式,避免使用引起QT间期延长的药物,并保持正常的电解质平衡,避免呕吐和腹泻等可能引起低钾血症的情况。还应避免基因型特异的触发因素,如针对LQT1的竞技运动与紧张和针对LQT2的声音刺激等。目前LQTS患者治疗目标可分为两种:通过β受体阻滞剂或左心交感神经切除术来减少交感神经兴奋以及通过植入型心律转复除颤器(implantable cardioverter defibrillator,ICD)适时终止致命性心律失常[5]。
需提醒的是,现代社会随着人们接触电子游戏的机会日渐增多,由此诱发LQTS患者发作心脏事件时有发生。Lawley等[16]在2019年就报道过LQTS及儿茶酚胺敏感性室性心动过速患者于电子游戏过程中发生室性心动过速及晕厥。笔者跟踪随访的病例也有过年轻患者自述在床上玩游戏时发作晕厥的情况。未来网络电子游戏等触发室性心律失常导致晕厥发作的情况可能会越来越普遍,应引起心律失常患者尤其是对交感神经兴奋异常敏感的LQTS和儿茶酚胺敏感性室性心动过速患者的高度警惕并加以避免。
4.1 β受体阻滞剂
自1970年以来,β受体阻滞剂已成为LQTS患者预防心脏事件发作的一线用药。除非有禁忌证,β受体阻滞剂是当今对LQTS患者的首选治疗。ACC/AHA/HRS在制定对室性心律失常和猝死防治指南中均建议对有症状的LQTS患者使用β受体阻滞剂(Ⅰ类推荐)。对无症状的LQTS患者,若其QTc≥470 ms,也应使用β受体阻滞剂(Ⅰ类推荐);若其QTc<470 ms,β受体阻滞剂则为Ⅱa类推荐[17-18]。对于QTc正常但携带致病基因突变者也建议使用β受体阻滞剂(Ⅱa类推荐)[2]。
在不同种类β受体阻滞剂中,普萘洛尔最为常用,通常每日剂量2~4 mg/kg,每次10 mg,每日3次起始,每隔5~7天加量5 mg,直至患者所能耐受的最大剂量。普萘洛尔的优势在于其脂溶性可通过血脑屏障,长期使用的耐受性较好,但其缺点是对哮喘患者禁忌使用。因此,纳多洛尔作为替代亦较常用,通常每天1 mg/kg,分两次使用[19]。国内推荐使用的是普萘洛尔和长效美托洛尔缓释片,运动试验时的峰值心率下降30%可能是β受体阻滞剂到达最大合适剂量的指标之一。
4.2 钠通道阻滞剂
β受体阻滞剂对LQT3患者较其他亚型疗效差。对有症状的LQT3患者可能需加用钠通道阻滞剂。根据指南规定,包括美西律、氟卡尼或雷诺嗪等钠通道阻滞剂,可作为QTc>500 ms的LQT3患者的补充治疗,以缩短其QT间期[2,17]。近年来又发现美西律对治疗LQT3之外的部分其他LQTS亚型患者也有效。如Bos等[20]报道,美西律在12例欧美LQT2患者中可使67%(8/12)患者QTc缩短40 ms以上。本组也报道过美西律治疗LQT8的研究,1例反复发作TdP伴晕厥的女性儿童患者,经美西律治疗后,室性心律失常发生明显减少。同时,笔者还发现其房室传导阻滞的发生亦明显减少,且进一步的功能研究提示,美西律通过降低晚钠电流,使得平台期内向电流减少(LQT8的发病机制为平台期L型钙通道功能获得性突变引起的钙内流增多,引起平台期延长),从而相对缩短了复极时程。提示美西律可有效治疗包括LQT3和LQT8这类以复极期内向电流增大为细胞电生理基础的疾病[21]。
4.3 电解质补充剂
维持LQTS患者的血钾在一个相对较高的水平可能有益,尤其是对LQT2患者,一般建议血钾水平>4 mmol/L,因为LQT2患者容易血钾低。建议患者在稳定期时,可在服用足够剂量普萘洛尔的前提下,同时服用门冬氨酸钾镁(潘南金)和/或补达秀等;在发作期可予以含更高浓度K+的极化液,将血钾快速升高至目标值[6]。
4.4 左心交感神经切除术
自主神经系统在心脏活动中发挥重要作用,自主神经紊乱是心律失常发生的重要机制。左心交感神经切除术对高危患者或对不耐受β受体阻滞剂的患者,可降低其心律失常发作的风险。此外,高危婴幼儿无法进行ICD植入时,也可选择左心交感神经切除术[17]。切除范围包括左星状神经节的下1/3和左侧T2~T5交感神经链,从而减少去甲肾上腺素对心脏的影响,此术式可减少LQTS患者晕厥的发作频率,又尽最大可能避免了Horner综合征。
4.5 ICD治疗
ICD是预防LQTS患者心源性猝死的有效手段,特别是对于猝死生还患者,指南推荐β受体阻滞剂联合ICD治疗( Ⅰ 类推荐)[2]。对接受β受体阻滞剂后仍反复发作晕厥的LQTS患者,ICD治疗作为Ⅱ a类推荐;对QTc>500 ms且携带KCNH2或SCN5A致病突变的无症状患者,亦建议安装ICD(Ⅱ b类推荐);对无症状且未开始使用β受体阻滞剂的LQTS患者,不推荐ICD治疗[17]。对一些高危个体,如QTc>500 ms的女性LQT2、JLNS或Timothy综合征患者,可预防性地使用ICD。
5 aLQTS
aLQTS可由多种原因引起,其中药物是最常见的诱因。理论上,任何一种延长复极的药物都可引起QT间期延长,可表现为U波增高,TU波异常,T波电交替或心率减慢等,继而引起TdP。引起aLQTS的常见药物包括抗心律失常药物、抗精神药物、大环内酯类抗生素和抗组胺药等。多种脑血管疾病也可导致aLQTS。也有观点认为所谓aLQTS可能是一些携带沉默突变的cLQTS患者,在某些药物的作用下破坏了复极储备,出现了外显的症状[6],随后陆续也有报道一些药物引起LQTS的相关基因[22-23](表3)。临床上需仔细筛查有无继发引起QT间期延长的病因,去除诱因后复查心电图是否有QT间期的缩短。
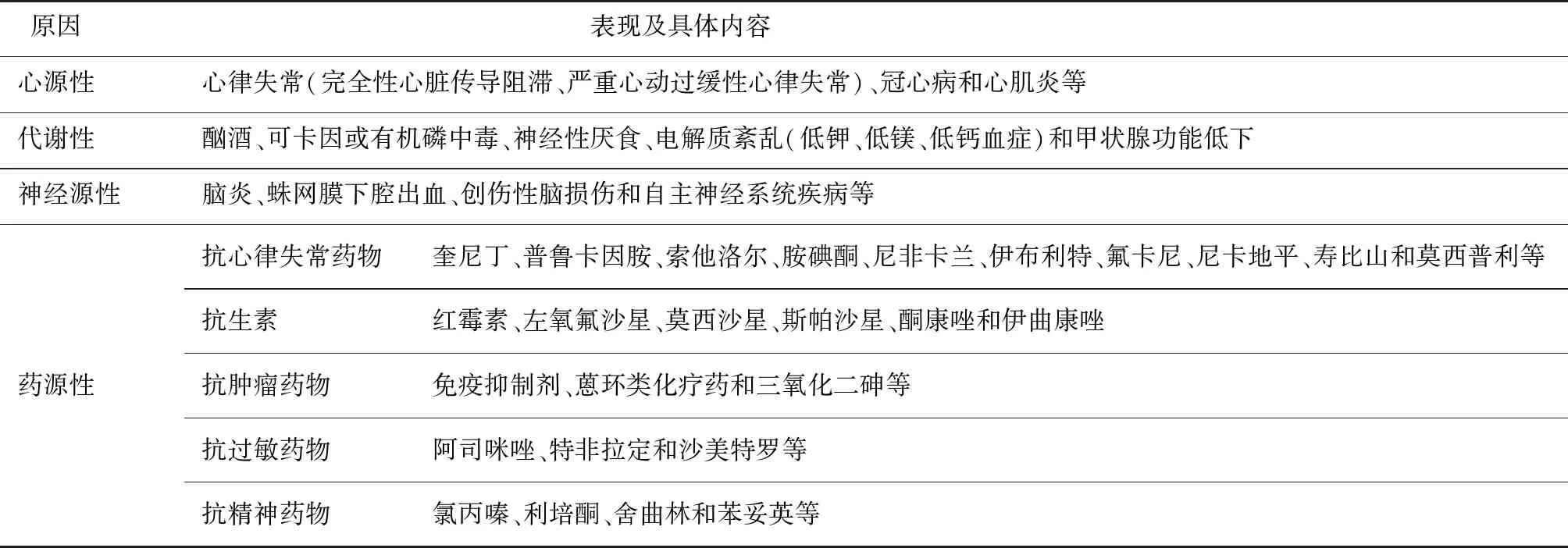
表3 aLQTS的常见原因
在COVID-19流行期间,对一系列药物(氯喹、羟氯喹、阿奇霉素、洛匹那韦/利托那韦、法拉匹韦、瑞德西韦和中药等)进行了COVID-19的临床试验评价。Szekely等[24]报道了第一例使用氯喹后QTc由462 ms延长至627 ms,并导致TdP的病例。停用氯喹及其所有可能延长QTc的药物6 h后记录到典型间歇依赖性TdP,患者当时无症状,提高心率及对症处理后,QTc逐渐恢复正常。
氯喹和羟氯喹同属于喹啉衍生物,均可通过抑制IKr通道导致QTc延长,但目前普遍认为单独使用时引起QTc显著延长的概率较小,倾向于在LQTS患者中不推荐阿奇霉素与氯喹或羟氯喹联用。β受体阻滞剂作为cLQTS一线用药,在和氯喹、羟氯喹或洛匹那韦/利托那韦等合用时,需监测β受体阻滞剂血药浓度并调整其剂量。此外,COVID-19患者中有10%~15%伴有继发性细菌感染需使用抗生素治疗。大环内酯类和氟喹诺酮类抗生素均可导致QTc延长,在LQTS患者中应避免使用这些药物,而选择对QTc无影响的抗生素[25]。
目前使用的抗冠状病毒鸡尾酒疗法,可能会增加药物相关LQTS的风险,进而可能恶化成药物相关TdP,甚至药物相关心源性猝死。那么,什么人感染COVID-19后更容易发生重症死亡呢?据Giudicessi等[26]报道,在美国COVID-19患者的地域分布研究中发现,非洲裔美国人占确诊COVID-19患者的26%,而死亡人数却占到全部COVID-19死亡的43%(非洲裔美国人占美国总人口的13.2%——本文作者加注)。这其中除了诸多文化和社会经济因素外,对新型冠状病毒感染的遗传易感性不容忽视。已有证据支持SCN5A-S1103Y多态性会增加药物相关LQTS和药物相关心源性猝死风险,而S1103Y在非裔人群的发生率为8%,其他种族的发生率为0.003%~0.400%[27-28]。S1103Y引起的晚钠电流增加可被低氧/酸中毒加重,进而增加室性心律失常和心源性猝死的风险。一些降低心脏复极储备的非遗传因素,如低钾、延长QTc的药物和/或结构性基础心脏病,可进一步加剧这种促心律失常的潜在风险。正常生理情况下(pH值6.9~7.1),S1103Y Nav1.5钠通道功能正常,晚钠电流不超过峰钠电流的0.5%。然而,当继发于低氧/低通气的呼吸性酸中毒发生时(pH值6.6~6.8),S1103Y钠通道会产生一个促心律失常和类似于LQT3的持续晚钠电流(约为峰钠电流的5%)[29]。临床观察到的许多COVID-19患者明显的低氧可能会促使S1103Y携带者更容易发生上述的促心律失常情况。
提到这类遗传多态的潜在影响,就不能不提在中国人群及亚洲人群广泛存在的另外一个多态SCN5A-R1193Q。R1193Q多态在中国汉族人群中的发生率是4.6%~7.9%(本组待发表数据为7.5%),在日本人群是1.7%~10%,在韩国人群是2.7%~8.2%,在西方人群是1%[30]。有多个离体研究证明R1193Q可能增加晚钠电流,但临床并未观察到R1193Q携带者出现LQT3表型。最近Kroncke等[31]报道,当R1193Q在人诱导多能干细胞(induced pluripotent stem cell,iPSC)分化的心肌细胞纯合表达时显示可增加晚钠电流,延长动作电位时程,并频发触发性细胞跳动。加入三磷酸磷脂酰肌醇(PIP3)可逆转上述过程。而PIP3与氧化应激反应呈负相关[32]。因此有理由推测,正常生理状态下,PIP3浓度正常,R1193Q不产生晚钠电流;但当炎症/低氧等因素使氧化应激增加时,PIP3可降低,进而使R1193Q携带者的晚钠电流增大,动作电位时程和QT间期延长,心律失常和心源性猝死的风险就会增加。类似于前面提到的S1103Y情形,推测COVID-19患者明显的低氧也可能会促使R1193Q携带者更易发生上述的促心律失常情况,故理论上推测R1193Q携带者罹患COVID-19后发生心律失常的风险可能会高。当然最终的结论还需进一步的研究来证实。
基于目前的COVID-19药物治疗研究,有学者[25-26]总结出了当使用可能延长QTc的药物治疗COVID-19患者时,可采取的风险评估流程,以避免引起aLQTS,降低药物相关TdP/药物相关心源性猝死,如图2所示。
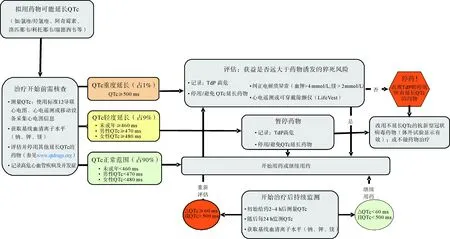
图2 为降低aLQTS的风险避免发生药物引起的尖端扭转型室性心动过速/心源性猝死,在使用1种以上延长QTc药物治疗COVID-19患者时,建议采取的风险评估流程(基于参考文献[25]和[26]修改)
6 未来的研究方向
使用患者体细胞来源的特异iPSC技术进行心肌分化已被广泛用于LQTS的致病机制研究、药物筛选和组织工程等许多方面,且所得结果比其他异源表达系统或转基因疾病模型更加接近于患者情况。此外一些新型治疗方案如规律性重复短回文序列簇相关Cas9(clustered regularly interspaced short palindromic repeats-associated Cas9,CRISPR/Cas9)系统基因修复、RNA干扰及过表达载体等,可在iPSC模型基础上进行评估。CRISPR/Cas9是一种精确高效的基因编辑技术,可由对照iPSC生成等位基因的突变细胞,也可从遗传上纠正一个突变iPSC,从而消除表观遗传学差异或未知遗传学修饰作用,后者可能会在致病突变的研究中引入表型差异。如Garg等[33]于2018年报道,利用CRISPR/Cas9技术纠正了LQTS疾病表型而制成了等位基因对照,从而证实一个原来定义为“意义不明变异”其实是一个能致病的突变。
然而iPSC模型仍有一定的缺陷,如iPSC分化心肌细胞效率有待提高,且所得心肌细胞分化成熟度亦不完全,其形态学上与人体心肌细胞相比还存在较大差异等,这使得将其转化为临床治疗仍有许多障碍。CRISPR/Cas9系统基因修复需在分裂细胞S期起作用,而人心肌细胞主要是静止细胞;心肌结构完整,无法移植外源心肌前体细胞;组织工程技术尚不能将纠正突变后的iPSC构建为形态功能完整的心脏组织。如何突破上述限制将是未来研究的方向和重点。
