一例难治性儿童局灶节段性肾小球硬化并文献复习
2021-06-06李倩倩唐玉英安虹瑾邴丽娟郑东丽穆善善
李倩倩,唐玉英,安虹瑾,邴丽娟,郑东丽,穆善善
(兰州大学第二医院甘肃省儿童医院小儿风湿免疫科,甘肃 兰州 730030)
1 病历资料
患儿,女,13岁,因“间断双下肢水肿1月,加重3d”于2020年6月3日收住入院。患儿于入院前1月无明显诱因双下肢出现重度水肿,伴晨起腰部酸困,无腰痛,无肉眼血尿,无尿频、尿急,无发热,无咳嗽,无心悸、胸闷,无恶心、呕吐,遂就诊于我院门诊,查速检生化示:白蛋白22.5g/L,甘油三酯1.91mmol/L,低密度脂蛋白7.05mmol/L,血常规示:血小板348×109/L。尿蛋白2+,24h尿 蛋白定量2.82g/24h,免疫球蛋白IgG3.03g/L。诊断为“肾病综合征”,并规律服用“强的松60mg qd、骨化三醇0.25g qd、双密达莫50mg tid”等药物,服药后患儿双下肢水肿较前减轻,但未完全缓解,仍伴腰部酸困。患儿入院前10d常规在当地医院查白蛋白24g/L,甘油三酯7.5mmol/L,低密度脂蛋白5.77mmol/L,血常规示:血小板425×109/L。免疫球蛋白IgG3.03g/L。24h尿蛋白定量2.2g/24h,尿蛋白2+。患儿于入院前3d出现咳嗽、咳痰,痰为淡黄色,易咳出,伴鼻塞,无咽痛,无发热、寒战,无恶心、呕吐,双下肢水肿较“感冒前”无明显变化,于当地医院查24h尿蛋白定量3.9g/24h,为求进一步治疗,遂就诊于本院,以“肾病综合征”收住。患儿此次病程中,精神状态好,饮食睡眠可,大便正常,小便色黄,有较多泡沫,近期体重无明显增减。患儿预防接种按时有序接种,没有食物、药物过敏史。G2P2,足月顺产,生长发育同适龄儿;患儿父母近亲婚配(表兄妹近亲结婚),体健,患儿姑姑有“肾炎”病史(具体不详)。入院查体:T为36.7℃,P为85次/min,R为20次/min,BP为117/81mmHg,W为51kg,发育正常,营养中等,神志清,精神反应无异样。全身皮肤黏膜无皮疹及黄染,浅表淋巴结未扪及明显肿大。头颅五官端正无畸形,双眼睑轻度浮肿,口唇红润,咽轻度充血,扁桃体无肿大,颈软无抵抗,呼吸平稳,听诊双肺呼吸音略粗,未闻及干、湿性啰音。心腹查体阴性,双下肢轻度凹陷性水肿。神经系统查体未见阳性体征。辅助检查:尿常规示:蛋白3+,隐血1+,红细胞63.8/ul,11.5/HPF。尿肾功:尿微量白蛋白2491.1mg/L;24h尿蛋白定量8.71g/24h;生化:白蛋白20.8g/L,总胆固醇8.10mmol/L,甘油三酯5.31mmol/L。入院诊断:(1)肾病综合征(肾炎型);(2)急性支气管炎。入院后治疗方案为:泼尼松60mg qd,贝那普利10mg qd,氯沙坦钾片50mg qd及补钙、补钾等对症治疗,同时给予抗炎、抗病毒、利尿、纠正低蛋白血症等对症治疗2周后复查尿蛋白3+,24h尿蛋白6.47~9.65g/24h,血压较高,波动在140~160/80~110mmHg,考虑对激素不敏感,于2020年6月10日行肾活检,病理回报示:符合局灶节段性肾小球硬化症,非特殊型(FSGS,NOS)(如图1~4所示)。眼耳检查未见异常。调整治疗,加用他克莫司胶囊1mg起始量口服,后检测药物浓度后调整至1mg bid,检测尿常规、尿肾功及尿微量白蛋白,患儿尿蛋白仍提示大量蛋白尿,并血压高,眼睑轻度水肿。患儿父母有近亲结婚史(表兄妹近亲结婚),且患儿姑姑有“肾炎”病史(具体不详),结合患儿肾穿,存在遗传性肾病高风险因素,故进行基因检测。2020年8月3日行遗传病医学外显子组基因测序回报:检测到受检者携带PTPRO基因一个纯合变异[c.792_793del,p.(Phe265fs)]。
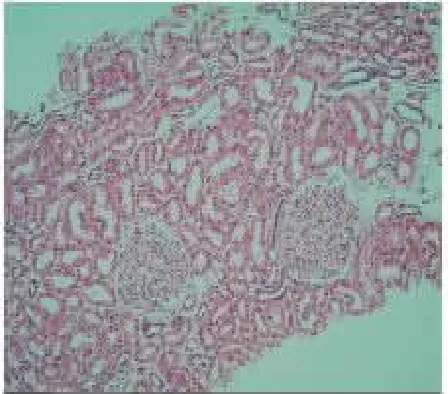
图1 HE×100

图2 PAS×400
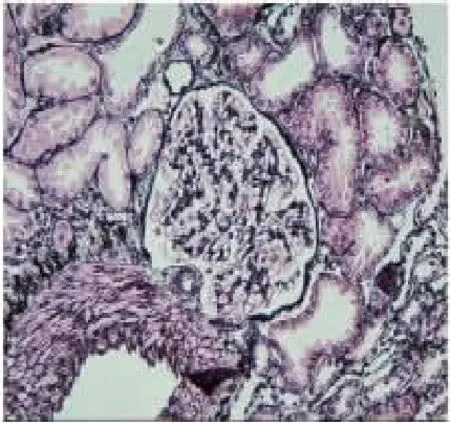
图3 PASM×200

图4 MASSON×400
2 讨论
原发性局灶节段性肾小球硬化(Focal segmental glomerulosclerosis,FSGS) 是儿童难治性肾病综合征常见的病理类型之一,局灶性肾小球节段性硬化是其病理特征[1]。临床表现为肾病综合征,伴有肉眼或镜下血尿、高血压[2],大约占儿童肾病综合征的10%~20%[3],FSGS预后比较差,50%的患儿在诊断[4]后在5~10年内进展为晚期慢性肾脏疾病。FSGS是终末期肾病(ESKD)[5]的主要病因之一。儿童和成人的FSGS发病率逐年在上升[6,7]。
近几年人们对于FSGS的病理形态学特征进行了深入的研究[8]。FSGS特征性病理是足细胞的损伤,在“足细胞病”中被认为是最具有代表性的病理类型,其组织病理学特征为肾小球节段性硬化,伴有或者不伴肾小球毛细血管内泡沫细胞形成、粘连。D’Agati等人[9]将FSGS分为五种病理类型:塌陷型(collapsing variant)、细胞型(cellular variant)、顶部型(tip variant)、门周型(perihilar variant) 以及经典型(not otherwise specified)。研究发现FSGS的病理类型不同,其临床表现及发病机制不同。FSGS最重要的发病因素是足细胞应力发生不可逆的改变,最后导致足细胞脱离,甚至凋亡。
另外,遗传因素在FSGS的发病中占重要地位。从遗传学的角度,通常把FSGS分为家族性FSGS和散发性FSGS两类。家族性FSGS具有遗传的异质性[10-12],研究证实是由多基因致病的一种单基因病,近几年国内外的学者研究了FSGS家系连锁分析以及定位克隆,发现了明确导致FSGS的致病基因,如NPHS1、NPHS2、TRPC6、INF2、ACTN4及PLCE1等基因,再次证明了家族性FSGS存在明显的遗传异质性;这些基因分别编码裂孔膜上和足细胞的重要蛋白podocin、nephrin和α 辅肌动蛋白4,肌动蛋白重要调控因子,磷脂酶Cεl及瞬时感受器电位蛋白C6。这些基因发生突变以后,通过影响细胞的动态调整、信号传导及骨架结构等多方面的发病机制,使足细胞受到损伤,最终导致FSGS。本例中检测到受检者携带PTPRO基因的一个纯合变异,该变异为移码突变(预计会导致所编码的蛋白质,自第265位氨基酸Phe开始发生移码,最终导致翻译提前终止)。预计该变异会导致所编码的蛋白质发生截短,从而丧失其正常生理功能。查HGMD数据库,没有该变异的相关文献报道,且HGMD数据库暂未收录该基因的移码和无义突变致病的文献报道;ESP6500siv2_ALL、千人基因组(1000g2015aug_ALL)和dbSNP147数据库均未见该移码突变的收录。综合患儿病史及遗传因素,认为该变异为可疑的致病变异。PTPRO基因作为蛋白酪氨酸磷酸酶PTPs家族成员之一,是新近发现的一个热门的、潜在的抑癌基因,是一种重要的信号通路媒介,参与细胞的增殖、代谢、分化、细胞与细胞间通讯、基因转录以及细胞存活等信号通路。多项研究表明在多种恶性肿瘤中,如肝癌、肺癌、白血病、乳腺癌及食道癌的病理组织中都存在该基因的低度表达。如果PTPRO基因发生致病变异,会引起肾病综合征6型,遗传方式为常染色体隐性遗传。患儿的父母往往携带致病变异。携带致病变异基因的父母,每次生育,子女成为患病者的几率均有25%。患儿父母的其他一二级亲属,也具有携带同样致病变异基因的风险。
国际儿童肾病研究(ISKDC)的一份报告显示,在FSGS患儿中,对激素敏感和对激素耐药的分别占3%和47.5%[13]。其中高达80%的原发性FSGS对激素是耐药的[14]。大部分激素耐药的FSGS患儿会进展为终末期肾脏疾病[15]。因此,对于FSGS患儿的治疗仍存在很大的挑战。近几年提倡的“精准医学”,在预防、诊断和治疗疾病中有重要意义,其核心理念是针对每个人的基因、生活方式和环境的不同,通过“精准医学”来更为准确地针对特定人群、特定疾病进行疾病的预防、诊断靶向治疗,并疗效预测疾病的预后。FSGS患儿中约10%~20%存在明确的家族史,应详细地询问病史并调查家族史,而且对于家族性FSGS的患儿,要调查世代至少要查3代。对于儿童时期起病的FSGS患儿,多呈常染色体隐性遗传(AR),这些患儿的父母亲常有近亲结婚史,遗传方式为常染色体隐性遗传(AR),本例患儿父母为近亲婚配,符合常染色体隐性的方式遗传特点。合理利用基因检测技术诊断疾病,能够发现治疗靶点,并能预测治疗效果,判断疾病的预后。除PLCEI等少数基因突变导致的肾病综合征对激素治疗敏感外,大多数遗传性FSGS患儿对激素治疗不敏感,反应比较差,因此,在治疗前很有必要进行基因诊断,可以预测药物的疗效,这样可以避免激素或免疫抑制剂不必要的治疗,减少药物的副作用。查明致病基因,可以为部分罕见单基因突变所致的遗传性FSGS患儿,提供精准的治疗靶点。该例患儿基因检测为罕见的单基因突变所致的家族遗传性FSGS,目前尚未见PTPRO基因纯合突变导致的FSGS的病例报道,亦未有有效的治疗方案,故治疗方案在摸索中,在采用足量激素(泼尼松)联合免疫抑制剂(他克莫司)治疗无效,且出现了难以控制的高血压、水肿、低钙血症及感染等并发症,患儿就诊于北京儿童医院,治疗方案调整为:停用泼尼松、他克莫司,继续口服贝那普利、氯沙坦钾片及维生素D、碳酸钙等药物治疗,患儿血压逐渐将至正常,水肿消退,复查24h尿蛋白维持在0.39g/24h(2020.10.19),血肌酐稳定在正常范围,离子钙水平及25羟维生素D水平恢复正常,患儿一般情况可,治疗处于临床部分缓解,目前患儿还需对疗效进行长期观察。
