结晶样视网膜色素变性中与CYP4V2基因相关的致病突变
2021-06-04杨极韦春玲张娟胡竹林焦康为
杨极,韦春玲,张娟,胡竹林,焦康为
0引言
结晶样视网膜色素变性(Bietti crystalline corneoretinal dystrophy,BCD)是一种常染色体隐性遗传性眼病。此病早期病情隐匿,患者往往因视功能损害不明显而未及时就诊,临床主要特征为眼底视网膜内白色结晶样物质沉积,脉络膜血管硬化,或伴有角膜边缘营养不良[1]。BCD在欧美人群中较少见,其致病基因为CYP4V2,有学者报道我国BCD致病基因频率为0.005,群体患病率约为1/24000[2]。大部分患者在10~40岁出现视力减退、夜盲、中央暗点等症状,随后发展为周边视野缺失和中心视力缺损,通常在40或50岁后视力下降至法定盲[3-4]。2015年孟晓红[5]对184个BCD家系中215例患者进行大规模临床检查和基因诊断后,充分揭示了中国人群BCD临床特点和基因突变谱,目前已知的致病性CYP4V2基因突变位点已达82个,故对患者进行临床诊断的同时进行基因筛查有利于增加疾病的精确诊断率,为进一步深入研究BCD的发病机制和为相关治疗提供依据。本研究对2个BCD家系进行Sanger测序,筛查患者的致病突变,现将结果报道如下。
1对象和方法
1.1对象描述性研究。收集2019-01/09就诊于云南省第二人民医院眼科门诊并确诊为BCD的先证者2例。从先证者出发,详细询问其家族遗传史,绘制家系图,并收集家族中其他成员纳入研究。共搜集BCD家系2个,家系1有明确的家族遗传病史,先证者及其哥哥,妹妹均患有BCD。家系2中先证者为散发病例,其父母已故。其中2例先证者均为女性。本研究遵守《赫尔辛基宣言》,经云南省第二人民医院伦理委员会批准,参与本研究的成员均签署知情同意书。
1.2方法
1.2.1检查方法对2例BCD先证者进行了详细的病史、家族史询问,同时进行了全面的眼科检查。临床检查包括裸眼视力、色觉、最佳矫正视力(BCVA)、裂隙灯下眼前段及眼底检查、眼底照相、眼部光学相干断层成像技术(OCT)检查、眼底荧光造影检查。
1.2.2基因检测签署知情同意书后,使用EDTA抗凝管采集先证者及参与研究的家族成员静脉血5mL,用基因组DNA提取试剂盒提取外周血DNA。提取的DNA需满足样本浓度均>50ng/μL,D(260nm)/D(280nm)为1.8~2.0,DNA样本-20℃冰箱保存[6]。根据美国国家生物技术信息中心提供的CYP4V2基因序列和参考文献[4]设计引物,利用PCR技术扩增目标DNA序列。采用传统50.0μL PCR扩增反应体系,加入DNA模板1.0μL,上、下游引物各1.0μL,2×EasyTaq R CR SuperMix 25.0μL,加入超纯水22.0μL。94℃预变性5min,94℃变性30s,50℃~65℃退火30s,72℃延伸1min,共反应35个循环,最后72℃延伸5min。操作步骤及流程严格按照云南省第二人民医院实验中心生化实验室要求进行。
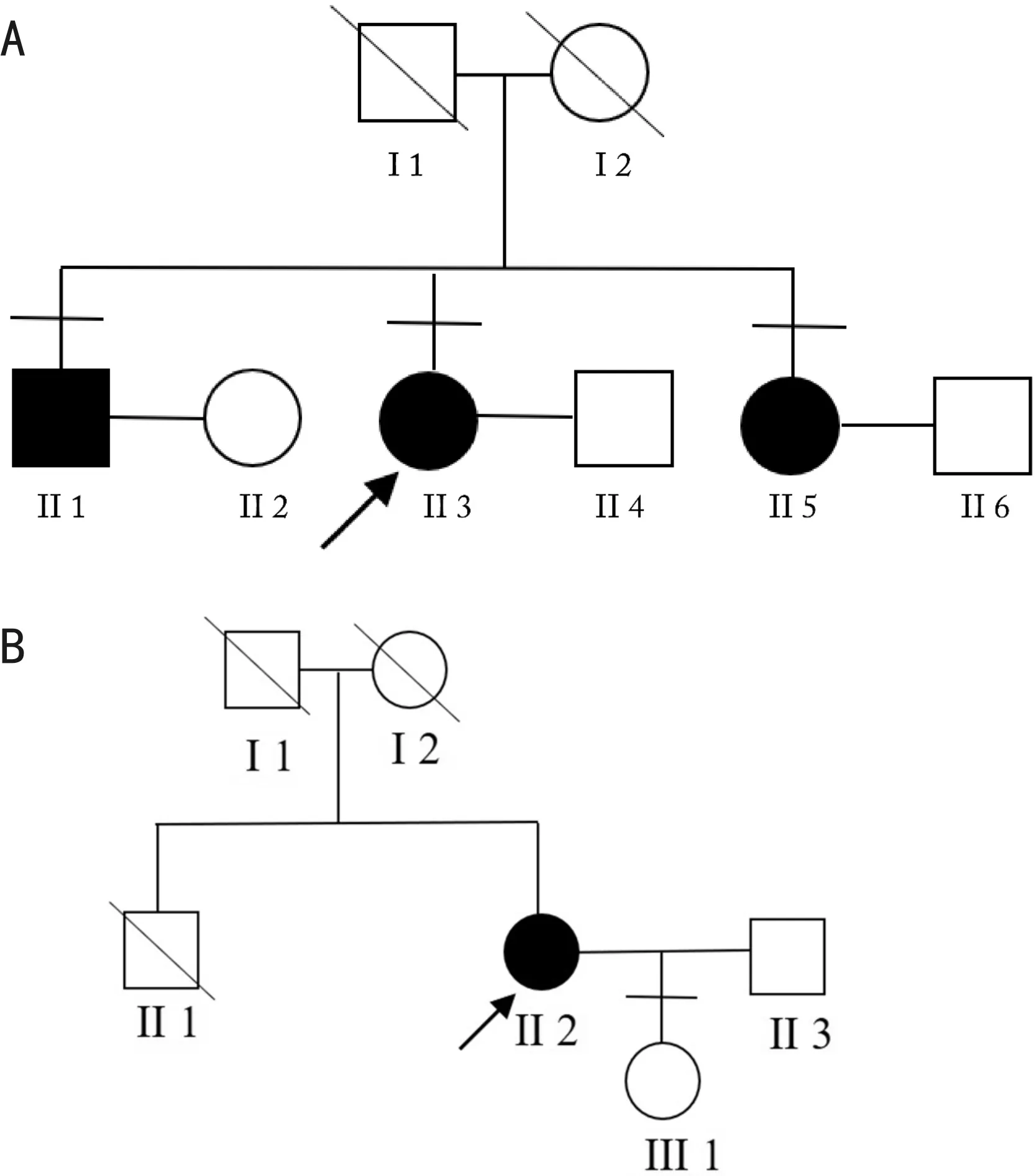
图1 患者家系资料 A:先证者1家系图;B:先证者2家系图。
通过dbSNP,ExAC Browser,the 1000 GenomesProject,Genome Aggregation Database等数据库获得变异的最小等位基因频率,运用 Mutation Taster、SIFT、PolyPhen2、GERP++、REVEL等生物信息学蛋白功能预测软件来进行危害性预测,并制作三维蛋白模型结构[7-9]。采用同源序列多重比对,结合OMIM、NCBI等数据库[8],利用UGENE
软件分析突变位点在不同物种间的保守型。依据美国医学遗传学与基因组学学会(ACMG,2015)分级标准对突变位点进行致病性判断[10]。
2结果
2.1 BCD患者临床特征先证者F1 II3(图1A),女性,33岁,主诉为“双眼渐进性视力下降1a”,BCVA:OD:0.05,OS:0.2。眼前节均未见明显异常,眼底可见大量密集黄白色结晶样颗粒物质沉积,后极部视网膜广泛脱色素至颜色变淡(图2A~F)。色觉检查:红绿色弱。后极部OCT提示:椭圆体带及色素上皮层萎缩,仅黄斑区少许残留,在萎缩区可见大量细小颗粒样物质均匀堆积,其下脉络膜毛细血管及中血管层相应萎缩,清晰可见大血管轮廓(图2G、H)。荧光造影检测可见结晶颗粒未显影(图2I、J)。哥哥F1 II1,男性,44岁,BCVA:OD:0.02,OS:0.05,妹妹F1 II5,女性,31岁,BCVA:OD:0.1; OS: 0.02,均可见眼底大量密集黄白色结晶样颗粒物质沉积。
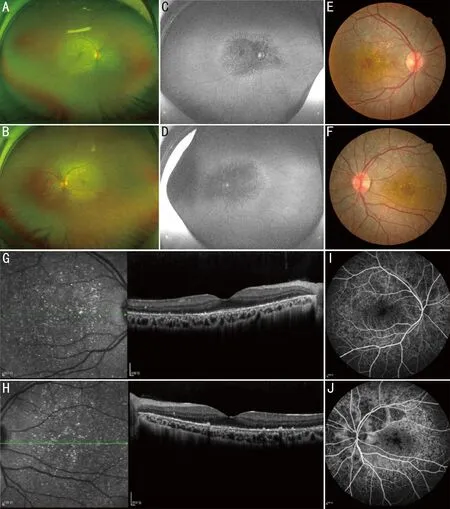
图2 同一患者影像学资料 A、B:广角眼底照,不同程度的结晶样物质沉积,伴视网膜色素上皮层萎缩及色素沉着;C、D:自发荧光可见视网膜结晶样物质沉积区域低荧光;E、F:眼底照,不同程度的结晶样物质沉积;G、H:OCT检查椭圆体带及色素上皮层萎缩,仅黄斑区少许残留,在萎缩区可见大量细小颗粒样物质均匀堆积,其下脉络膜毛细血管及中血管层相应萎缩,清晰可见大血管轮廓;I、J:荧光造影可见结晶样物质荧光遮蔽。
先证者F2 II2(图1B),女性,41岁,主诉为“双眼渐进性视力下降7a”,BCVA:OD:0.05; OS:0.12。眼前节均未见明显异常,眼底见不同程度的结晶样物质沉积,伴视网膜色素上皮层萎缩及色素沉着。女儿F2 Ⅲ 1,视力正常。
2.2 Sanger测序结果对先证者F1 II3和其近亲属F1 II1,F1 II5的外周血DNA进行Sanger测序后,检测出CYP4V2基因上存在c.802-8_810del17insGC的纯合突变(图3A~C)。根据ACMG指南(PVS+PS1+PM2),该变初步判定为致病性变异。PVS:该变异为零效变异,可能导致基因功能丧失。PS1:HGMD数据库已有该位点的致病性报道。PM2:在dbSNP,ExAC Browser,the 1000 Genomes Project等数据库中为低频变异。同时保守性分析显示此突变位于不同物种间氨基酸高度保守的位置。
由于先证者F2 II2的父母、哥哥已故,无法进行家系共分离验证,对先证者和其女儿的外周血DNA进行全外显子测序后,检测出先证者CYP4V2基因上存在和F1家系一致的c.802-8_810del17insGC杂合突变,同时F2 II2还存在c.219T>A(p.F73L)突变(图3D~E)。依据ACMG指南(PS1+PM2),c.219T>A(p.F73L)为疑似致病性变异,PS1:HGMD数据库已有该位点的致病性报道。PM2:在正常人数据库中为低频变异。同时生物信息学蛋白功能预测软件MutationTaster、GERP++、PolyPhen2均预测此突变为有害。保守性分析显示c.802-8_810del17insGC,c.219T>A(p.F73L)突变位于不同物种间氨基酸高度保守的位置(图4A、B)。3D蛋白预测模型提示c.219T>A(p.F73L)突变影响蛋白质的局部构像(图4C、D)。先证者F2 II2无BCD症状的女儿发现CYP4V2基因上存在c.219T>A(p.F73L)杂合突变(图3F)。
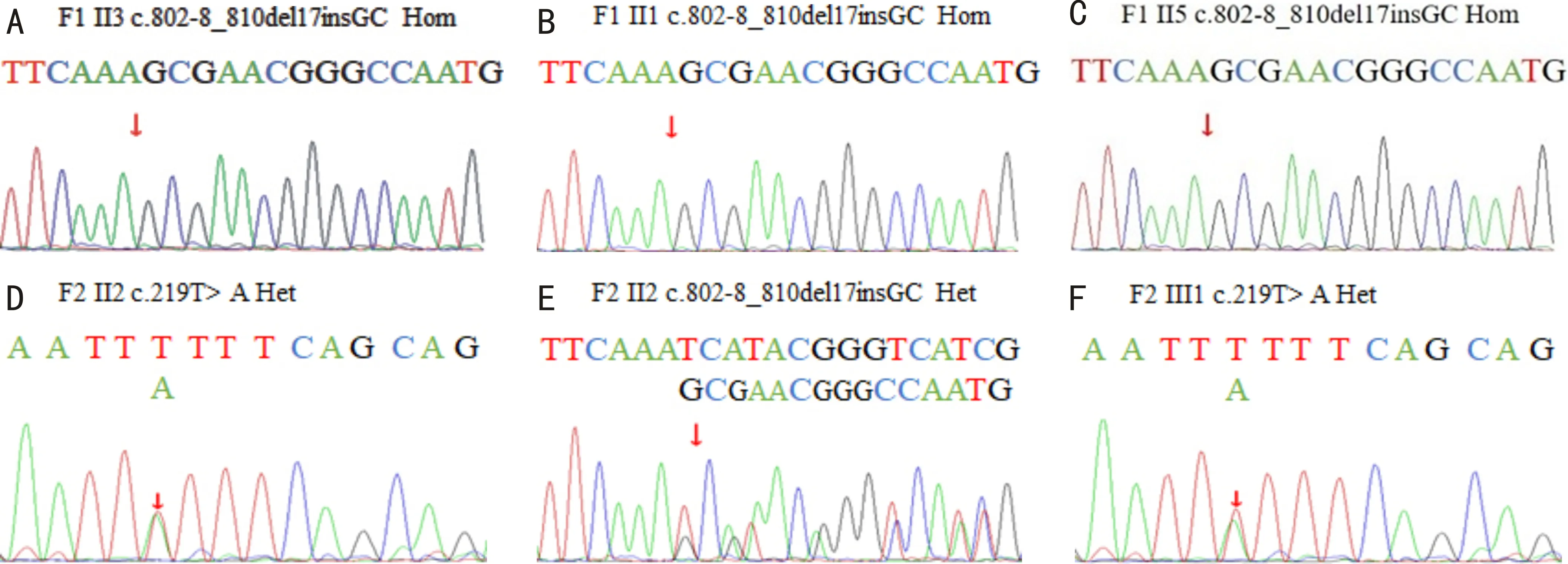
图3 Sanger测序结果 A~C:家系1中II3,II1,II5检测出CYP4V2基因上存在c.802-8_810del17insGC的纯合突变;D~E:F2 II2检出c.219T>A(p.F73L)和c.802-8_810del17insGC杂合突变;F:F2 III1检出c.219T>A(p.F73L)杂合突变。
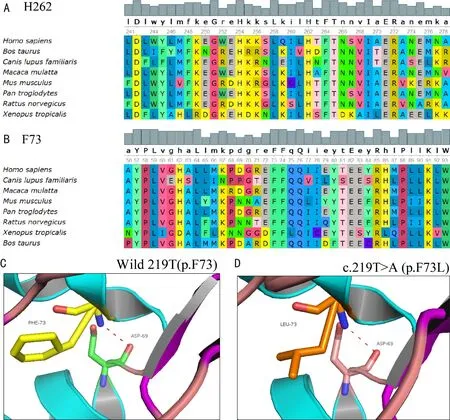
图4 保守性分析和蛋白三维结构预测结果 A、B:保守性分析显示c.8028_810del17insGC(H262),c.219T>A(p.F73L)(F73)突变位于不同物种间氨基酸高度保守的位置;C、D:3D蛋白预测模型提示c.219T>A(p.F73L)突变影响蛋白质的局部构像。
3讨论
BCD(OMIM#210370)是一类常见的常染色体隐性遗传性视网膜变性疾病,在1937年由Bietti医生首次报道,其在视网膜色素变性疾病中比例高达18.5%,在中国人群中发病率远高于欧美人群[5, 11]。BCD的致病基因为CYP4V2,位于4q35-qter,由11个外显子组成,编码525个氨基酸,属于细胞色素氧化酶P450(CYP450)超家族[12]。目前CYP4V2导致BCD发病的机制研究还未完全明了,有研究表明CYP4V2参与脂肪酸代谢,BCD患者血清三酰甘油、总胆固醇、低密度脂蛋白胆固醇、高密度脂蛋白胆固醇和正常人群存在差异,但CYP4V2突变对全身各组织器官影响较小,推测其发病方式可能为基因突变影响了视网膜色素上皮细胞的脂质代谢而致病[13]。BCD具有高度遗传异质性,至今已发现的BCD致病突变位点达82个,绝大部分为错义突变、缺失以及无意突变,涉及所有11个外显子区域,对BCD患者进行进一步,尤其是致病基因CYP4V2相关突变位点筛查明确病因具有重要意义[14-15]。
本研究应用Sanger测序技术对4例BCD患者和1例BCD患者健康的女儿进行了基因检测。通过基因检测,我们发现在CYP4V2基因上存在c.219T>A(p.F73L),c.802-8_810del17insGC突变。本文研究的2个BCD家系,先证者均具有明显的BCD表现,其中F1 II3和其近亲属II1,II5均具有多年的夜盲病史,伴有视力下降,眼底存在典型的结晶样物质沉积,并检测到c.802-8_810del17insGC纯合突变。先证者F2 II2眼底可见结晶样物质沉积,同时存在c.219T>A(p.F73L),c.802-8_810del17insGC复合杂合突变,先证者F2 II2的女儿发现CYP4V2基因上存在c.219T>A(p.F73L)杂合突变,但先证者F2 II2女儿未出现BCD的症状。
本研究发现的c.802-8_810del17insGC突变为中国人群中CYP4V2基因最常见的突变方式,在CYP4V2基因突变比例中占62.6%,此突变可导致氨基酸编码终止进而影响蛋白功能的发挥。Lai等[3]研究报道了c.802-8_810del17insGC突变与表型间的关联,发现突变眼电图上可显示低Arden比,并表现出不可记录的暗适应FERG,以及不可记录的30Hz闪烁ERG。Gekka等[16]比较了2例CYP4V2突变患者的临床特征,发现纯合缺失/插入突变(c.802-8_810del17insGC)的患者均出现了视野受损。1例患者的视杆、视锥细胞ERG振幅严重降低。相反,另1例患者虽然有复合杂合子突变,临床表现轻微,视锥和视杆细胞的ERG振幅未受到明显影响。但是发病年龄和病程对视网膜功能改变相关因素的影响未见报道,不存在明显的基因型-表型关联。虽然表型与基因型之间存在较大差异,但表型的严重程度与疾病的病程有关,发病越早,严重表型持续时间越长,本研究中F1 II1已有夜盲病史10余年,患者目前44岁,已双眼失明。
先证者F2 II2检测到的c.219T>A(p.F73L)突变在2014年由Yin等[17]首次报道,此突变在健康人群中未被报道,在dbSNP,ExAC Browser,the 1000 Genomes Project等数据库中为低频变异,而73号位点的苯丙氨酸在多物种间均为一个高度保守的序列,三维蛋白结构预测也可见此突变影响了局部蛋白质的二级结构,进而影响了蛋白质的疏水性和稳定性。CYP4V2基因编码跨膜氨基酸与18个α螺旋以β折叠和卷曲的形式构成跨膜结构,此结构可与亚铁血红素相识别。错义突变可影响α螺旋与卟啉环的结合,进而导致该结构失去功能。CYP4V2基因的错义突变一般可影响蛋白跨膜段,干扰CYP4V2蛋白与膜的整合,在酶的活性位点干扰其与卟啉环的配位[18-19]。本研究再次证明了c.802-8_810del17insGC和c.219T>A(p.F73L)突变在中国BCD人群的发病中扮演重要角色。
但是,由于BCD的高度遗传异质性,对其了解仍然很少,基于通过研究大样本不同BCD患者的遗传特征,进一步阐明其致病的分子机制,不但提高该疾病诊断的准确率,对解释BCD疾病发生发展的本质和探索其基因型和表型之间的分析就显得尤为重要,同时可为开发该病个性化的基因治疗奠定基础。
