H 2O2/乙腈体系下MgO催化环己酮Baeyer-Villiger绿色氧化合成ε-己内酯的研究
2021-06-03朱倩倩靳海波郭晓燕何广湘马磊张荣月谷庆阳杨索和
朱倩倩,靳海波,郭晓燕,何广湘,马磊,张荣月,谷庆阳,杨索和
(北京石油化工学院化学工程学院/燃料清洁化及高效催化减排技术北京重点实验室,北京102617)
引 言
ε-己内酯是一种重要的新型聚酯单体和有机合成中间体,其开环聚合所得的聚己内酯具有良好的生物相容性、生物降解性、渗药性、热塑性并且无毒,常用作人工合成组织、药物释放载体、医用涂层的生产等,且在临床上得到广泛应用,同时还可与交联淀粉共混改性制备具有良好力学性能的可降解材料[1-5]。Baeyer-Villiger(B-V)氧化反应是将脂肪族酮氧化为酯,将环状酮氧化为内酯的反应,在有机合成中起着重要作用[6-8],ε-己内酯可由B-V氧化环己酮制备。目前,工业上主要使用有机过氧酸作氧化剂制备ε-己内酯,该方法会产生酸性废弃物对环境造成污染,并且过氧酸易于分解,具有一定的危险性[9-10];氧气氧化法常以醛类作为共氧化剂,但反应过程中副产大量有机酸,且不易分离[11-13];而生物氧化法虽具有高效性、专一性,且反应条件温和、无污染,但是技术成本高、稳定性差、效率低,仍停留在实验室阶段[14-15]。因此,从绿色安全的氧化剂及高效的催化剂入手开发环己酮绿色氧化新工艺具有非常大的经济与社会效益。
相较于过氧酸氧化,采用双氧水氧化副产物为水,清洁无污染。Olszówka等[16-17]使用双氧水作为氧化剂、镁铝类水滑石为催化剂进行了环己酮氧化合成ε-己内酯的研究,并研究了镁铝配比及其结晶度等对催化性能的影响,发现当Mg/Al=5.62时,ε-己内酯具有最高收率(50%),结晶度较低的水滑石催化剂具有较高的选择性,从而满足了绿色化学的要求。Pillai等[18-19]以乙腈为共氧化剂与双氧水共同参与反应,其中乙腈不仅起到溶剂的作用,而且乙腈与过羟基阴离子物质(HOO—)反应,生成了过氧缩酰亚胺酸,进一步氧化被活化的环己酮。Yang等[20-21]制备了Mg、Sn、Al和W等金属氧化物催化剂,经800℃煅烧得到锡钨摩尔比为2的复合金属氧化物,催化效果最佳,75℃下反应10 h,环己酮转化率可达88%。Ruiz等[22]将镁铝比为4∶1的水滑石催化剂用于B-V氧化催化环己酮反应,具有良好活性。清洁高效合成己内酯已日益成为一种趋势,而对其反应机理的探索也具有一定理论意义。Pillai等[18]研究发现环己酮氧化过程中H2O2分子与环己酮共同被激活,反应遵循过氧缩酰亚胺酸路线。Ma等[23]研究了过渡金属氧化物为催化剂的环己酮B-V氧化反应,MoO3和WO3具有较高的催化活性,并提出了MoO3和WO3的反应机理。Corma等[24]认为反应过程中只有环己酮被激活,H2O2分子直接进攻羰基碳原子,反应为一步反应。由于H2O2绿色清洁,乙腈极性强溶于多种有机物与无机物,乙腈与环己酮和过氧化氢水溶液均互溶,可使反应体系很好混合,产生单一液相反应混合物,比两相反应更易于处理。因此,本文以沉淀法制备MgO催化剂用于H2O2/乙腈体系下B-V氧化环己酮合成ε-己内酯,系统考察了MgO制备条件对其结构及催化性能的影响,并对反应条件进行进一步的优化。使用在线原位红外光谱对反应过程实时分析,进一步验证了其反应机理。
1 实验部分
1.1 主要试剂与仪器
Mg(NO3)2·6H2O,分析纯,西亚试剂;Na2CO3,分析纯,国药集团化学试剂有限公司;环己酮、30%H2O2、乙腈、氯仿,分析纯,北京化工厂。
高低温平行反应仪WP-TEC-1020HC,西安华泰科思实验设备有限公司;气相色谱GC-2010,日本岛津公司;气质联用仪Agilent 7890B-5977A,安捷伦科技有限公司;原位红外ReactIR 15,瑞士梅特勒-托利多;马弗炉;烘箱。
1.2 催化剂的制备
称取60 g Mg(NO3)2·6H2O溶于200 ml去离子水,电磁搅拌下滴加质量分数为5%的Na2CO3溶液,沉淀完全后,继续滴加2 mol/L NaOH溶液调节pH,搅拌1 h后,在室温下静置老化24 h,用去离子水洗涤,将洗涤后所得白色沉淀在60℃烘箱中烘12 h,研磨后将其置于马弗炉内以5℃/min的升温速率升温至一定温度下煅烧,冷却至室温,得到所需MgO。
1.3 催化剂的评价及产物分析
向100 ml的三口圆底烧瓶中依次加入0.1 mol环己酮,2.5 g催化剂,50 ml乙腈溶剂,并加入80 ml双氧水,加热至60℃反应5 h,反应结束后,冷却1 h,抽滤分离催化剂,用氯仿萃取反应液中有机相,对萃取后的反应液进行分析。
样品分析:使用GC-2010气相色谱仪,HP-5色谱柱(50 m×0.200 mm),FID检测器,初始温度120℃保持3 min,10℃/min升温至240℃,保持15 min。
MgO催化环己酮的氧化反应的性能评价指标为环己酮的转化率(X)和己内酯的收率(Y),分别采用式(1)、式(2)计算。

式中,m环己酮为环己酮加入的质量,g;m总为反应液的质量,g;w环己酮为反应液中环己酮的质量分数;w己内酯为反应液中己内酯的质量分数。
1.4 催化剂表征及条件
XRD:日本理学株式会社SmartLab-9 Kw X射线衍射仪;辐射源为Cu靶,Kα辐射源(λ=0.15418 nm),管电压为40 kV,管电流为30 mA,扫描速率4(°)/min,扫描范围20°~80°。
SEM:日本电子株式会社JEOL JSM-7600超高分辨热场发射扫描电子显微镜;测试电压1.5 kV,放大倍数为2千倍和10万倍。
2 结果与讨论
2.1 催化剂制备条件优化
沉淀法制备的金属氧化物催化剂,其煅烧温度以及煅烧时间是影响催化剂性能的重要因素,图1是考察催化剂制备的煅烧温度及煅烧时间对反应效果的影响。从图中可以看出,环己酮转化率及ε-己内酯收率随温度的增加而缓慢增加,在600℃时环己酮转化率及ε-己内酯收率达到最大,分别为74.7%和67.1%,之后环己酮转化率及ε-己内酯收率随温度的升高而减少;煅烧时间在2 h之前,环己酮转化率及ε-己内酯收率随煅烧时间的增加而增加,2 h时环己酮转化率及ε-己内酯收率达到最高,分别是87.7%和68.1%,2 h之后则开始减少。在较高的煅烧温度条件及较长的煅烧时间下催化剂的活性出现降低。这是因为煅烧温度过高会造成催化剂中金属氧化物的结构改变,催化剂活性组分晶粒生长较快,形成晶粒尺寸变大(这与XRD表征结果一致),使得反应物分子在催化剂活性中心上的吸附与脱附变得困难,或使活性组分结构发生变化,活性位数量减少。因此,煅烧温度为600℃、煅烧时间为2 h为最佳条件。
2.2 催化剂表征
2.2.1 催化剂XRD表征 MgO是典型的立方晶相结构。所制备MgO催化剂XRD谱图如图2所示,400℃时由于煅烧温度较低,前体煅烧分解不完全,有非晶相MgCO3·3H2O存在,并未完全分解为MgO,500~800℃下催化剂特征衍射峰尖锐(36.94°、42.92°、62.30°、74.69°和78.63°),五个晶面分别为(111)(200)(220)(311)(222),且与国际标准图谱库JCPP中MgO(PDF#NO.45-0946)的特征衍射峰完全一致,由图可知随着煅烧温度的升高,衍射峰强度逐渐增大,说明催化剂结晶度也在增大。因此,MgO在500℃以上煅烧较为合适。

图1 不同制备条件对催化剂性能的影响Fig.1 Effect of different preparation conditions on catalyst performance
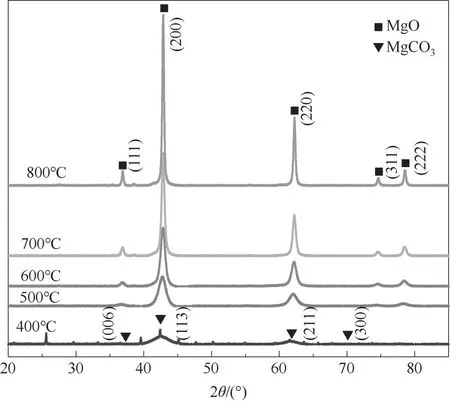
图2 不同煅烧温度下MgO的XRD谱图Fig.2 XRDpatterns of MgOat different calcination temperatures
由图1可知,随煅烧温度的升高,产率先增加后降低;但催化剂的接触角[25]由40°降低到28°,这也表明结晶度较低的MgO亲水性较强,有助于表面碱性中心上的H2O2接近和活化,而结晶度高的MgO的亲水性较差使得催化剂更易于与有机底物相互作用,随后进行非选择性转化[26-27]。根据Scherrer公式计算得到500~800℃MgO样品的晶粒尺寸依次为9.53、12.48、17.71、29.49 nm。这可能是因为温度较低时晶粒的生成较快,而晶核的生长较慢,所以晶粒尺寸较小;而在高温阶段晶粒生长速度要高于晶核形成速度,所以温度高有利于形成尺寸较大晶粒,并且在高温处理过程中,初级粒子的表面原子活性将进一步增强,有可能使粒子间的软团聚转化为硬团聚,晶粒长大。因此,煅烧温度对粒径大小的影响至关重要,在保证纳米粒子前体充分分解的基础上应降低锻烧温度[28-29]。
2.2.2 催化剂SEM表征 图3为不同煅烧温度下所制备MgO的SEM图像,由图3(a)~(d)可以看出MgO呈棒状,对比图3(e)~(h)可以看出500℃时棒状MgO表面几乎平滑,有少量结晶,600℃、700℃和800℃下MgO表面不再平滑,开始在表面出现类似球状颗粒,且粒径逐渐增大,这可能是因为随着温度升高,表面细小颗粒开始团聚,晶粒长大,图3(d)显示800℃时MgO出现烧结,与前述一致。而煅烧温度为600℃催化剂有最佳催化活性,说明MgO形貌对MgO催化活性有着重要影响。
2.3 反应条件的考察
2.3.1 催化剂用量的影响 催化剂用量对有机合成反应有一定的影响,图4为催化剂用量对反应效果的影响。从图4可以看出,ε-己内酯的产率随催化剂的量的增加而增大,在催化剂摩尔比为0.45∶1时,ε-己内酯产率达最大为71.6%,继续增加催化剂摩尔比时ε-己内酯产率无明显变化,分析认为MgO催化反应时,环己酮分子首先由反应体系进入MgO内部,经过MgO催化剂的活化中心活化后,再经过脱附与H2O2分子结合,当体系内反应底物量确定时,需要的催化剂量也一定,因此,当MgO用量增加到一定量时,转化率不再增加,所以最佳的催化剂摩尔比为0.45∶1。
2.3.2 溶剂用量的影响 溶剂在有机反应中有着重要作用,其中溶剂的偶极矩和介电常数是影响溶剂溶解溶质能力的两个重要因素。乙腈是一种优良的溶剂,具有较大的偶极矩和介电常数。本反应以乙腈作为溶剂,可以起到提高反应的传质速率,同时增加反应传热的作用,且乙腈还会与反应物环己酮结合,从而加快反应的进行,改变反应历程[30],反应机理部分对此做了详细讨论。
图5为溶剂用量对反应效果的影响,可以看出ε-己内酯随溶剂摩尔比先增加后减少,溶剂摩尔比为1∶12时ε-己内酯产率达到最大为73.6%,这是因为反应过程中有机相环己酮和无机相H2O2在乙腈溶剂中相结合,增大反应底物接触面积,提高传质速率,从而加快反应进行,当乙腈达到最佳量即1∶12时,再增加乙腈用量则会使反应物浓度降低,不利于反应的进行。因此,1∶12为最佳溶剂摩尔比。
2.3.3 双氧水用量的影响 环己酮氧化过程中,H2O2作为氧化剂其用量对产物ε-己内酯有较大的影响。由于H2O2氧化剂氧化能力较弱,通常需要过量使用。由图6可以看出,随H2O2量的增多,环己酮转化率在逐渐增大,当H2O2与环己酮摩尔比增大至1∶10时,ε-己内酯收率达最大为74.9%,但当其摩尔比在1∶10之后时,ε-己内酯产率有所下降。分析认为H2O2量增加时,体系水的量也相应增加,同时,双氧水呈弱酸性,这都会加剧产物ε-己内酯的水解,因此,在环己酮转化率增加的同时,出现了ε-己内酯产率下降现象,故H2O2与环己酮适宜的摩尔比为1∶10。

图3 不同煅烧温度下MgO的SEM图Fig.3 SEM imagesof MgOat different calcination temperatures
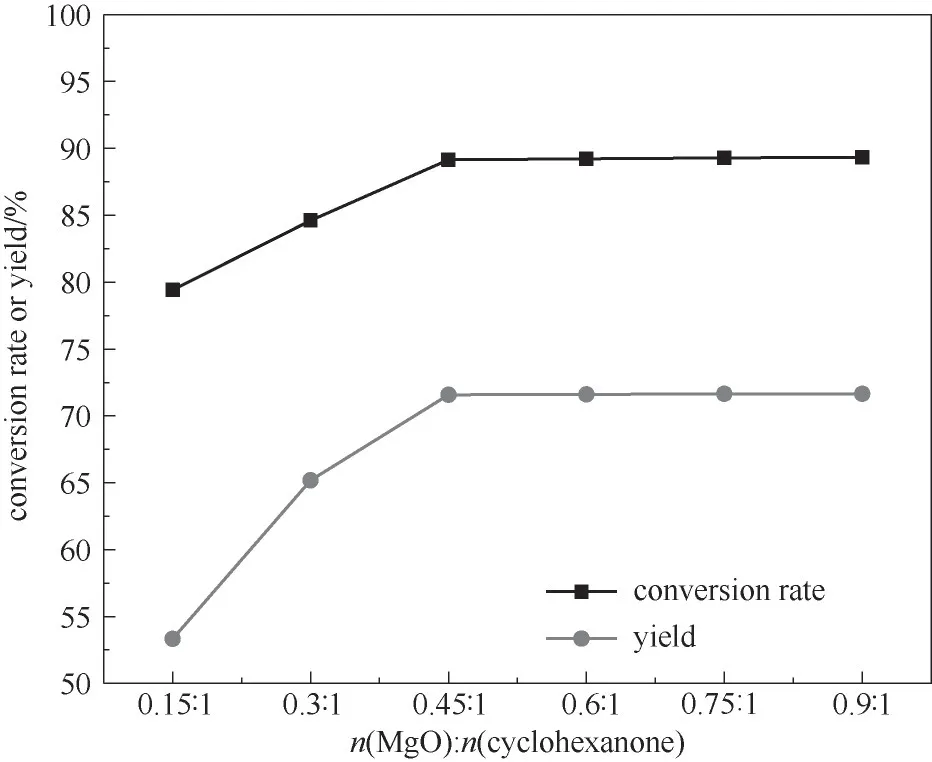
图4 催化剂用量对反应的影响(反应条件:环己酮30 mmol;30%(质量分数)双氧水35 ml;乙腈20 ml;60℃;5 h)Fig.4 The influence of the amount of catalyst on the reaction
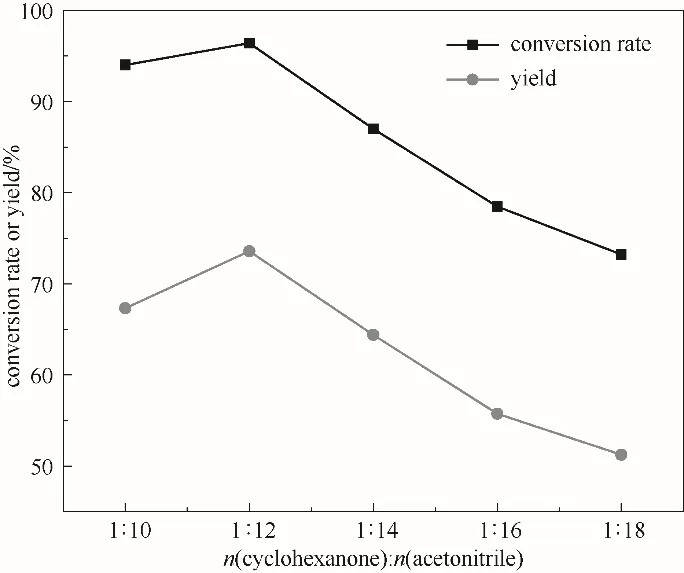
图5 溶剂用量对反应的影响(反应条件:环己酮30 mmol;30%(质量分数)双氧水35 ml;催化剂0.54 g;60℃;5 h)Fig.5 The effect of the amount of solvent on the reaction
2.3.4 反应温度的影响 考察反应温度对ε-己内酯收率的影响,图7表明,在70℃之前,随反应温度的升高,ε-己内酯产率增加,70℃时达到最大为77.0%,继续升高温度,ε-己内酯产率反而下降,这是因为较高的温度会加快H2O2的分解,同时,体系含水量增加,导致ε-己内酯水解增多,不利于反应进行,ε-己内酯产率降低,故选择最适宜的反应温度为70℃。
2.3.5 反应时间的影响 图8为反应时间对ε-己内酯产率的影响,在反应时间为6 h之前,环己酮转化率与ε-己内酯产率随反应时间的增加而增大,在6 h时环己酮转化率与ε-己内酯产率达到最大,分别为95.2%、83.1%;反应继续进行,虽然转化率继续增大,但收率缓慢减小。这是由于随着反应的进行,H2O2分解速率逐渐增加,同时反应产物ε-己内酯发生部分水解,ε-己内酯的选择性降低,收率不再增加,故适宜的反应时间为6 h。
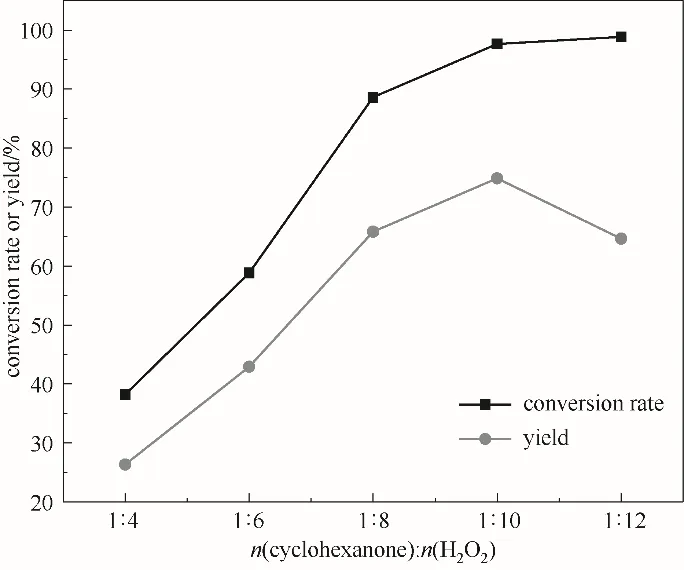
图6 双氧水用量对反应的影响(反应条件:环己酮30 mmol;乙腈15 ml;催化剂0.54 g;60℃;5 h)Fig.6 The effect of hydrogen peroxide on the reaction
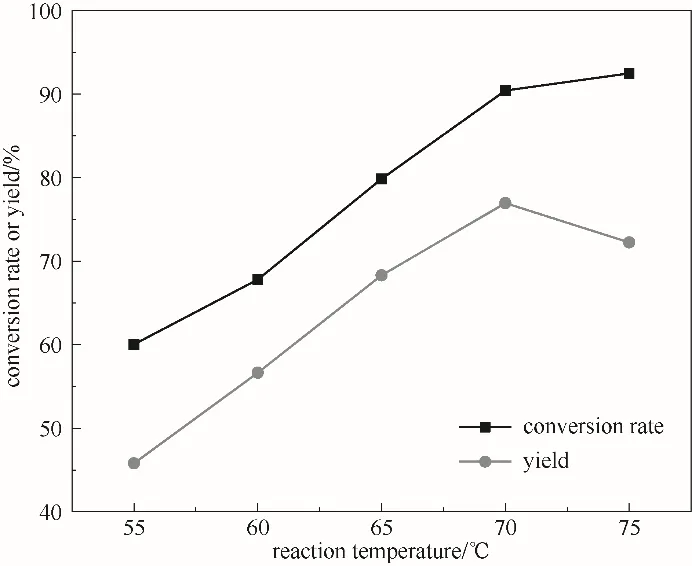
图7 反应温度对反应的影响(反应条件:环己酮30 mmol;30%(质量分数)双氧水30.7 ml;催化剂0.54 g;5 h)Fig.7 The effect of temperature on the reaction
2.4 副产物分析
在优化的反应条件下,当ε-己内酯产率最高为83.1%时,环己酮转化率为95.2%,选择性为87.3%,这说明环己酮氧化反应中还生成了副产物。为确定副产物组成,反应后的反应液用GC-MS进行定性分析,从而揭示反应的规律。环己酮随着反应的进行而不断减少,ε-己内酯不断增加;当ε-己内酯增加到一定程度后,由于体系中水含量较大,ε-己内酯发生水解,从而降低了反应的选择性。水解生成的6-羟基己酸在过量双氧水的作用下还可能进一步生成少量己二酸。另外,环己酮还可能发生加成反应生成2-环己烯-1-酮,进一步发生环氧化生成7-氧杂二环[4.1.0]庚烷-2-酮,如图9所示。
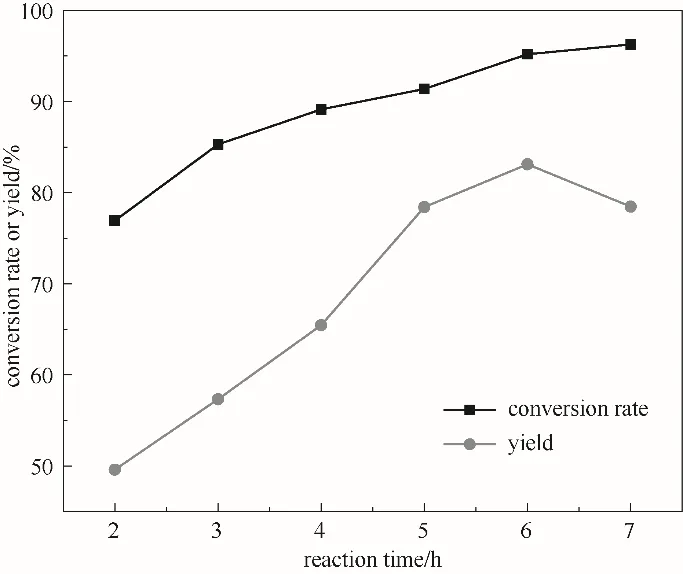
图8 反应时间对反应的影响(反应条件:环己酮30 mmol;30%(质量分数)双氧水30.7 ml;催化剂0.54 g;70℃)Fig.8 The effect of time on the reaction
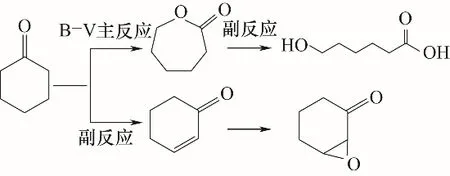
图9 环己酮B-V氧化副反应路线Fig.9 Cyclohexanone B-V oxidation side reaction route
2.5 反应机理
为了进一步探索其反应机理,使用在线原位红外光谱对反应机理进行验证。根据H2O2分子是否被活化,将反应机理分为以下两种。
①通过与催化剂的路易斯酸位点配位而使环己酮羰基活化,从而导致羰基碳的亲电性增强,促进H2O2分子的亲核进攻,接着,H2O2分子进攻环己酮羰基碳后形成Criegee加合物(a),(a)经过重排后形成ε-己内酯[26]。反应过程中仅环己酮的羰基碳被活化,而H2O2分子未被活化,此为一步反应路线,如图10所示。
②被催化剂活化的H2O2进攻氧化镁催化剂表面的碱中心,形成一种过氧中间体(a),提高了双氧水的亲核能力;由于该过氧中间体不稳定,迅速与乙腈结合形成过氧缩酰胺中间体(b);接着,在反应的第二步中,中间体(b)被催化剂的路易斯酸位点活化的环己酮的羰基进攻,形成类似Criegee的加合物(c);最后,中间体(c)经过重排,生成ε-己内酯和乙酰胺。反应过程中氧化剂H2O2和酮的羰基均被催化剂活化,使B-V反应加速,是一个两步反应路线,如图11所示。
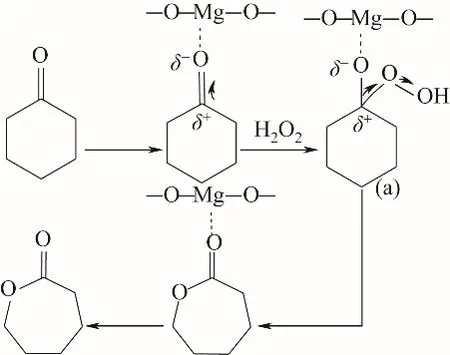
图10 路线Ⅰ:H2O2/乙腈体系下MgO催化环己酮B-V氧化反应机理(仅酮基碳被活化)Fig.10 RouteⅠ:MgO-catalyzed oxidation mechanismof cyclohexanone B-V in H2O2/acetonitrile system
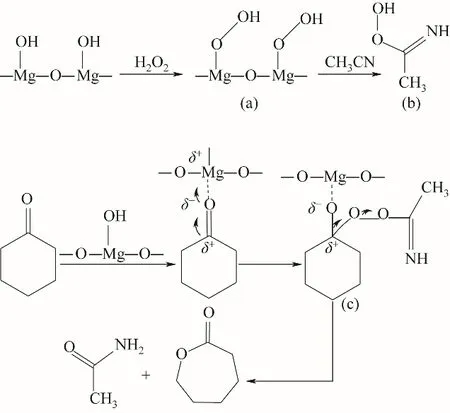
图11 路线Ⅱ:H2O2/乙腈体系下MgO催化环己酮B-V氧化反应机理(酮羰基与H2O2分子均被活化)Fig.11 RouteⅡ:MgO-catalyzed oxidation mechanismof cyclohexanone B-V in H2O2/acetonitrile system
为了验证H2O2/乙腈体系下MgO催化环己酮BV氧化反应机理,用原位红外对反应体系进行实时在线分析,测定的红外光谱如图12所示,可观察到1558 cm-1处过氧缩酰胺中C N伸缩振动及1410 cm-1处乙酰胺中C—N伸缩振动。路线Ⅰ中乙腈未参与反应,即反应过程中不会出现C N与C—N伸缩振动,而路线Ⅱ中H2O2与MgO催化剂所形成的中间体与乙腈结合会形成过氧缩酰胺,反应最后Criegee加合物重排后形成乙酰胺,分别有C N与C—N的伸缩振动。因此,验证了路线Ⅱ所示的反应机理。
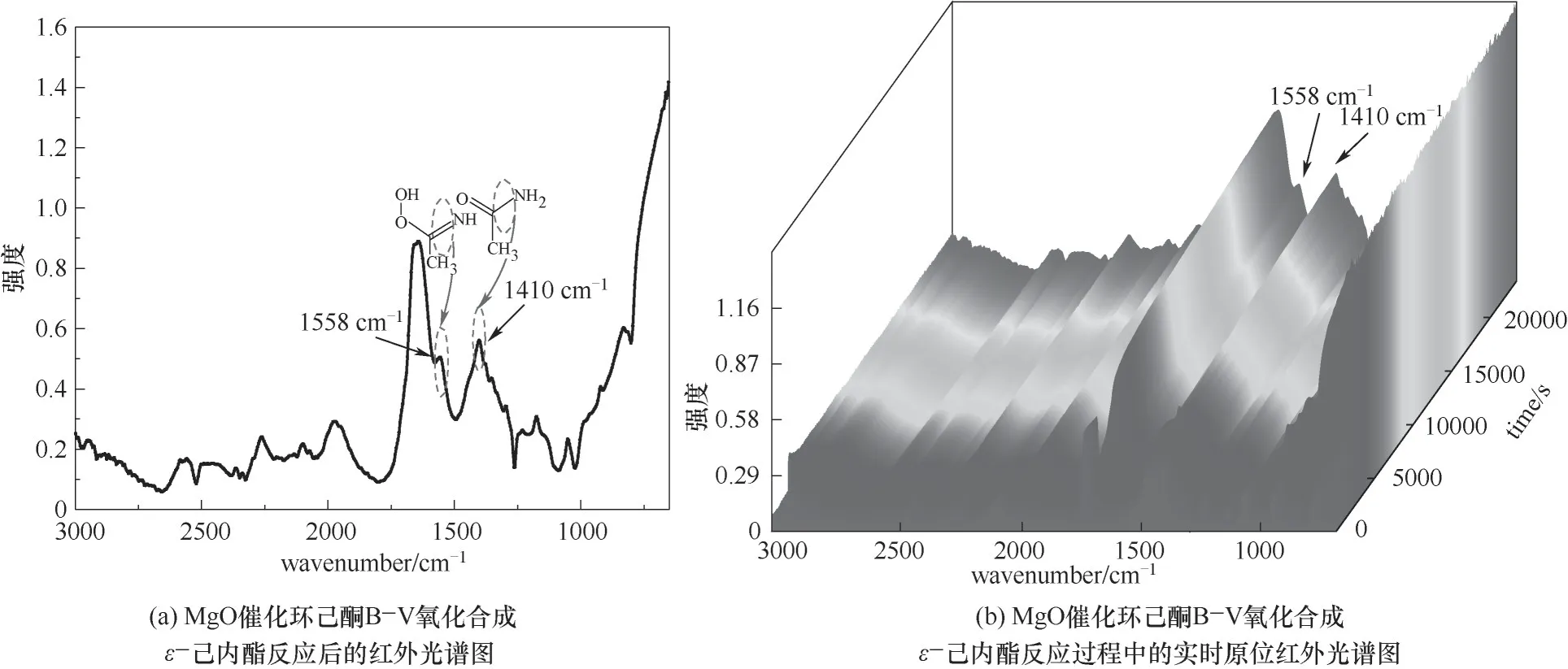
图12 MgO催化环己酮B-V氧化合成ε-己内酯原位红外光谱图Fig.12 In-situ infrared spectroscopy of MgOcatalytic cyclohexanone B-V oxidation toε-caprolactone
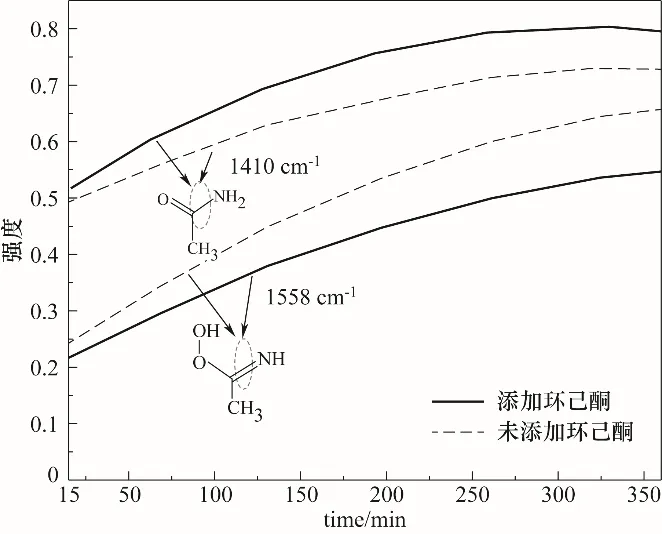
图13 MgO催化B-V氧化合成ε-己内酯中乙酰胺与过氧缩酰胺原位红外光谱峰强变化Fig.13 MgOcatalyzed oxidation of B-V to synthesizeεcaprolactone acetamide and peroxyacetal in-situ infrared spectrumpeak intensity changes
进一步对路线Ⅱ反应机理进行验证,原位红外测定不加环己酮反应时C N与C—N的伸缩振动与加入环己酮实验进行对比(图13为加入环己酮与不加环己酮反应6 h的C—N及C N伸缩振动峰的变化)。不加环己酮时出现1410 cm-1C—N,这是由于乙腈在H2O2存在的酸性环境下水解生成乙酰胺。从图13可以看出在1410 cm-1处C—N及1558 cm-1处C N伸缩振动峰都在增加。但两图对比可以发现,不加环己酮时1558 cm-1处C N伸缩振动峰比加入环己酮时峰强更强,而1410 cm-1处C—N伸缩振动峰比加入环己酮时峰强更弱,这说明形成的过氧缩酰胺中间体进行第二步反应与被活化的环己酮结合生成ε-己内酯,以使过氧缩酰胺的量减少而乙酰胺的量增多(即C N伸缩振动峰变弱,C—N伸缩振动峰增强),从而进一步验证了路线Ⅱ反应机理。
3结 论
以Mg(NO3)2·6H2O为原料通过沉淀法合成MgO,考察了催化剂的煅烧时间及煅烧温度对催化剂活性的影响,得到其最佳煅烧温度及煅烧时间分别为600℃、2 h,并对催化剂进行XRD及SEM表征分析,并用X射线衍射(XRD)、扫描电镜(SEM)进行了分析,由分析可知随温度升高MgO晶粒尺寸逐渐增大,在500~800℃范围内,MgO晶粒尺寸为9.53~29.49 nm,分析表明随温度升高,催化剂结晶度不断升高,其催化性能先增强后减弱;对反应条件进行优化,最佳反应条件为n(催化剂)∶n(环己酮)=0.45∶1,n(环己酮)∶n(乙腈)=1∶12,n(环己酮)∶n(双氧水)=1∶10,反应温度70℃,反应时间6 h,反应收率可达83.1%;分析了其反应副产物主要为由己内酯水解产生的6-羟基己酸及环己酮加成所生成的2-环己烯-1-酮;用在线原位红外光谱对反应进行实时在线分析,验证了其过氧缩酰胺反应路径。
