过渡金属催化CO2/H2参与的羰基化研究进展
2021-06-02华凯敏刘晓放魏百银张书南王慧孙予罕
华凯敏,刘晓放,魏百银,3,张书南,王慧,*,孙予罕,3,4,*
1中国科学院上海高等研究院,中科院低碳转化科学与工程重点实验室,上海 201203
2中国科学院大学,北京 100049
3上海科技大学物质科学与技术学院,上海 201203
4上海低碳技术创新功能型平台,上海 201620
1 引言
目前,大气中二氧化碳(CO2)的浓度已达到创纪录的水平,且在持续增长,这引发了全球气候变暖、海平面上升和海洋酸化等重大环境问题1。因此减少CO2排放成为国际社会的共识和高度关注的热点,也是国内产业升级和环境保护的重要指标2,3。另一方面,温室气体CO2也是一类理想的C1资源,具有储量丰富、可再生、廉价易得、无毒等优点。基于此,开发CO2利用技术以获得高附加值化学品4和燃料5等越来越受到关注,这既减少了碳排放,又促进了经济的可持续发展。
在CO2利用中,虽然CO2容易与强亲核试剂反应,它仍通常被认为是一种“惰性”分子,这是因为CO2已经处于碳的最高氧化态,本身化学性质稳定,其标准吉布斯自由能为ΔG = -394.38 kJ·mol-1,具有热力学稳定性和动力学惰性的特点。为解决CO2活化动力学上的挑战,近几十年来的基础研究持续关注于催化剂的开发,并取得了显著进步6-8。在过去的几十年里CO2利用被广泛地探索,将CO2转化为高附加值的化学品和能源产品取得长足的发展6,9。如图1所示6,根据CO2分子的还原水平和键形成过程(C=O键被C―H或其他C―元素键取代),CO2转化可分成三类。图1左侧CO2通过非还原性转化,生成诸如碳酸酯或聚碳酸酯等产品;右侧则是CO2被完全还原,生成甲烷(CH4)或饱和碳氢化合物。而中间一类转化结合了CO2还原和化学键形成,可获得一大类功能化分子,如甲酸、甲醇、醇、羧酸、酰胺、醛等大宗或精细化学品。这一类还原功能化无疑是最丰富多彩同时也最具价值的CO2转化之一。
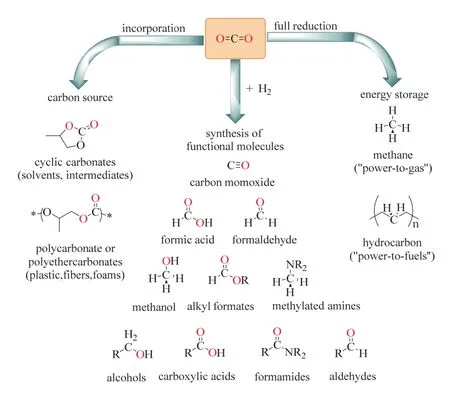
图1 CO2作C1源的资源化利用Fig. 1 Selective transformations of CO2 as C1 building-block.
为实现CO2的还原转化,通常需要通过热还原、光还原或电还原等过程提供较高的能量。CO2的光/电化学还原领域的相关工作也被广泛报道10,11。在CO2热催化的化学还原中常见的还原剂有H2、氢硅烷、氢硼烷等12。其中,可以从非化石资源中产生,如通过清洁能源光分解水生成的H2是最具应用前景的还原剂。CO2加氢常见产物有一氧化碳、甲酸、甲烷、甲醇、高级醇、烯烃、液态燃料等4,6,13-21,这些生成的功能产品实现了CO2/H2融入化学品“价值链”(图1,中间路径)中。其中,CO2催化加氢可以逆水煤气变换直接得到CO,也可以催化加氢得到CO替代品,如甲酸、甲醛等,这些CO替代品可以进一步分解得到CO。而CO是现代化工生产中不可或缺的大宗化学品,如可参与到羰基化反应中22。羰基化反应是指通过催化的方法,在有机化合物分子内,引入羰基及其衍生基团的一类重要反应。经典的羰基化反应是不饱和化合物(烯烃、炔烃)、卤代烃和甲醇等在亲核试剂的存在下与CO反应生成应用广泛的有机含氧化合物,如图2所示。
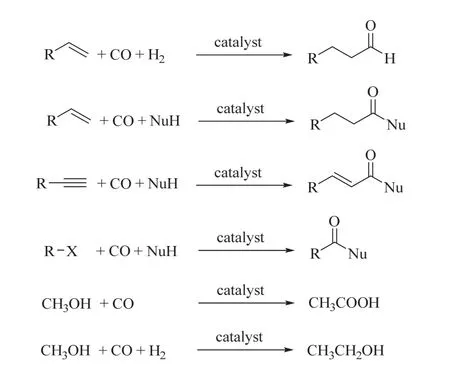
图2 CO参与的羰基化反应Fig. 2 The conventional carbonylation with CO.
因此,近年来,人们尤为感兴趣的是使用CO2/H2替代CO合成高附加值化学品23,以期在未来工业生产中实现CO2作为主要含碳化学品的来源。Leitner等在Science的Perspective中就描述了CO2/H2替代CO的前景,将CO2/H2形容为“初见不钟情”,这是因为CO2是“不情愿的”搭档(热力学稳定和动力学惰性),但在特殊催化剂(主要为过渡金属络合物)的“撮合”下,实现了CO2/H2“再见钟情”,参与到高价值化合物的合成中(如图3所示)24。
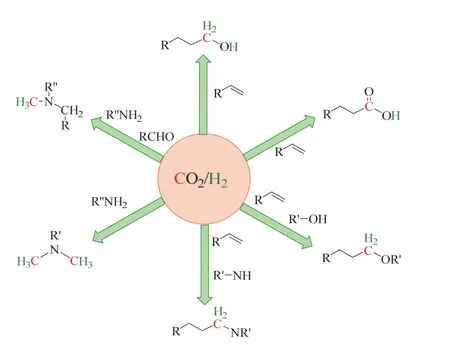
图3 Leitner等描述的CO2/H2替代CO的前景Fig. 3 The prospect of CO2/H2 as CO surrogate described by Leitner et al.
过渡金属催化CO2/H2的组合为替代CO提供了一种可能,为合成高价值的化学品创造了基础,潜力巨大。本综述对近年来CO2/H2参与的羰基化研究进展进行了简要的总结和评述。在此,我们将CO2/H2看成类似魂斗罗一样不断闯关的搭档,每闯过一个关卡即成功实现了一种羰基化反应,在这些关卡中会出现不同的障碍:氢化/氢解、反应条件苛刻、一般都涉及多个竞争反应、化学选择性低、区域选择性差、贵金属用量过高或催化性能低等,而化学家们通过给两位“魂斗罗”装备不同的“道具”(如卤代盐、离子液体、膦配体等)成功克服了通向目标产物道路上的“障碍和敌人”。
2 CO2/H2参与的烯烃羰基化反应
烯烃的官能团化是当今化学工业的重要基础。除了聚合和氧化外,使用CO进行烯烃羰基化反应是工业生产醛、醇、羧酸、酯、胺等高附加值和精细化学品的主要技术(如图4所示),对烯烃的高效羰基化也是有机化学研究中的重点课题和挑战之一25,26。

图4 CO参与的烯烃羰基化反应Fig. 4 The carbonylation of alkene with CO.
众多研究者对CO2/H2作CO替代品参与烯烃羰基化反应进行了广泛地研究和探索27,在过渡金属催化下成功实现了CO2/H2参与烯烃羰基化反应生成醛、醇、羧酸、酯和胺等28。在此,我们综述了以CO2/H2为CO替代品的烯烃羰基化反应的研究进展。根据已有的研究,在CO2/H2参与的烯烃羰基化反应体系中目前主要有四种策略(如图5):①钌催化的RWGS/烯烃氢甲酰化/氢化的多步串联反应生成醇,若将还原剂H2替代为醇,则可生成产物酯;②负载在二氧化钛(TiO2)上的金(Au)为代表的多相催化体系成功实现了多相RWGS/乙烯氢甲酰化/氢化制丙醇的反应;③铑催化的RWGS/烯烃氢羧化串联反应生成高级羧酸;④ CO2被聚甲基氢硅氧烷(PMHS)还原为CO,随后经铑催化的烯烃氢甲酰化选择性生成醛。这些催化体系通过巧妙的设计实现了CO生成和烯烃羰基化的高效耦合,同时不同的催化体系也存在着各自的优劣势。我们通过对不同烯烃羰化反应类型的分析,阐述该领域的研究进展。
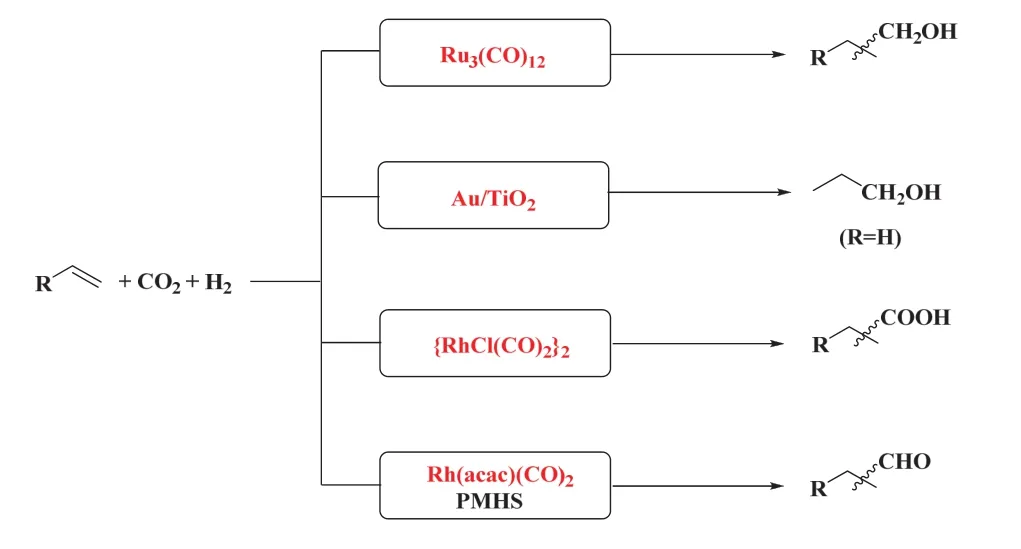
图5 CO2/H2参与的烯烃羰基化反应的策略Fig. 5 Strategies for carbonylation of alkenes with CO2/H2.
2.1 CO2/H2参与的烯烃氢甲酰化/氢化制醇
1993年,Sasaki课题组首次报道了均相催化RWGS,在CO2/H2氛围下,以十二羰基三钌(Ru3(CO)12)为催化剂,KI为添加剂,反应机理如图6a所示29。首先,CO2插入Ru―H键中(A),在H2辅助下脱去一分子水,生成羰基Ru络合物(B),随后B脱除CO,脱羰后的Ru进一步加氢生成Ru―H键,催化剂循环再生。但该体系中羰基Ru络合物(B)同时会生成副产物CH3OH和CH4等。次年,同课题组在上述催化体系的基础上,引入LiCl或双(三苯基正膦基)氯化铵([PPN]Cl)为添加剂,不仅使CO2高效选择性转化为CO,同时成功抑制了CH4和CH3OH的生成,反应温度也显著下降(图6b所示)30,为利用原位生成的CO参与羰基化反应提供了可能。基于Ru3(CO)12催化RWGS的特性,同一课题组于2000年将该体系拓展至烯烃氢甲酰化反应中,首次实现了CO2/H2参与的RWGS/烯烃氢甲酰化/氢化制醇的反应(如图7a所示)31。该催化体系使用四核羰基钌(H4Ru4(CO)12)为催化剂,LiCl为添加剂,反应在140 °C、CO2/H2(4/4 MPa)条件下进行。CO2首先经历RWGS生成CO;在H2和原位生成的CO参与下,烯烃发生氢甲酰化反应生成醛。但在高温条件以及具有显著氢化倾向的Ru存在下,反应产物不能停留在醛,而是进一步氢化生成醇,最终环己基甲醇收率可达88% (以环己烯为模板底物)。这项开创性的工作引起了广泛关注,众多科研工作者在此基础上深入研究了CO2/H2参与烯烃制醇的反应,以期开发出CO2为羰源的适宜工业化的烯烃氢甲酰化的新型催化剂。
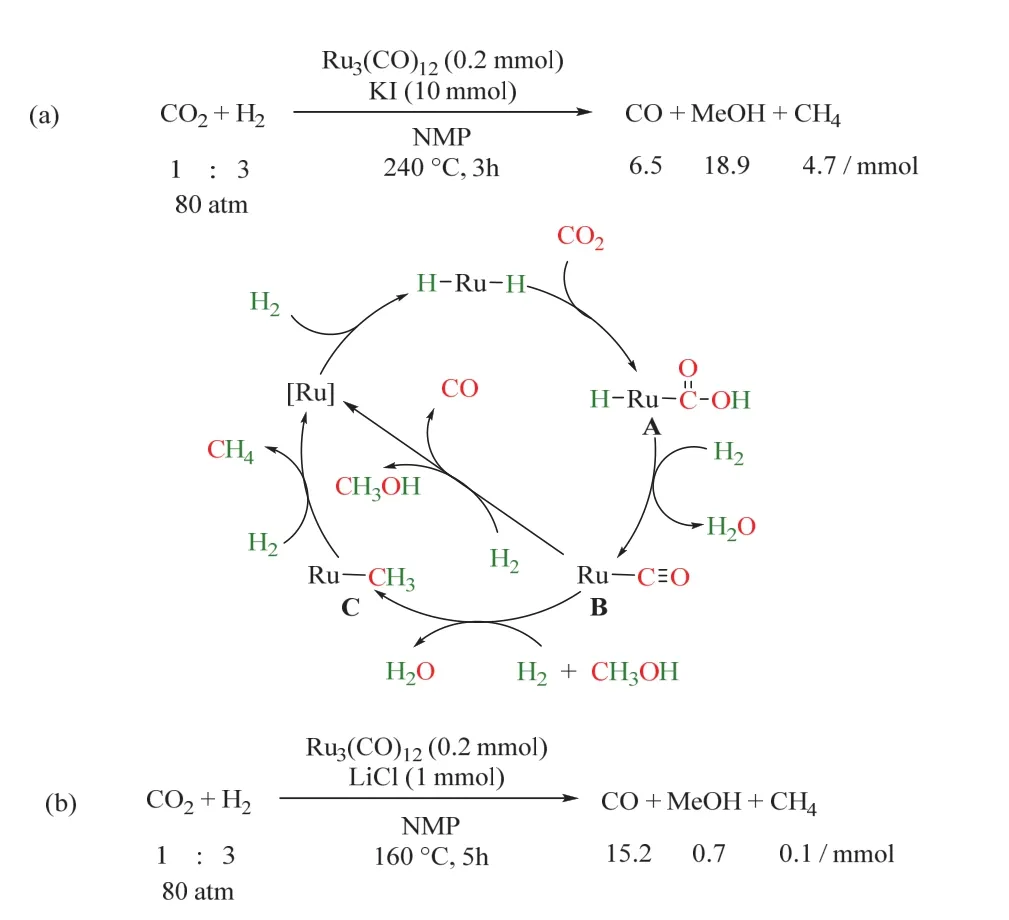
图6 Ru3(CO)12催化CO2逆水煤气变换为CO机理图Fig. 6 A plausible reaction mechanism of RWGSR.

图7 Ru催化的CO2/H2参与的烯烃羰基化反应制醇Fig. 7 Ru-catalyzed alkene carbonylation to produce alcohols with CO2/H2.
后续研究发现卤素添加剂对氢甲酰化反应也有着显著影响32-34,当没有添加剂卤代盐时,氢甲酰化反应不发生,仅得到烯烃氢化产物。金属催化剂前体中羰基不可或缺,不含羰基的常见钌金属盐无法催化这类反应。同时N或P配体的引入降低了反应性能,可能的原因是配体抑制了CO的生成。在相同的反应条件下,与传统的CO羰基化相比,CO2表现出相似甚至更好的性能。其反应循环机理如图8所示34,外圈经历RWGS生成CO,ESIMS表明该过程是由四核阴离子物种催化的。首先Cl-脱除[H2Ru4(CO)12]2-中的质子生成不含氢的络合物[Ru4(CO)12]4-,这是该过程的关键步骤;随后CO2配位生成[Ru4(CO)12(CO2)]4-,质子亲核进攻进一步的把配位的CO2转化成CO配体,形成[Ru4(CO)13]4-;后者在H2辅助下脱去CO,同时再生为[H2Ru4(CO)12]2-。原位生成的CO随后参与内圈的氢甲酰化反应。烯烃首先与催化活性中心[RuCl3(CO)3]-生成烷基钌络合物[R’RuCl2(CO)3],随后CO插入,在HCl辅助下脱去醛,催化剂[RuCl3(CO)3]-循环再生;最后,醛被Ru催化氢化生成醇。由图可以看出,卤素离子的主要作用是脱除质子,从而生成具有反应活性的催化中心羰基钌阴离子[Ru4(CO)12]4-。
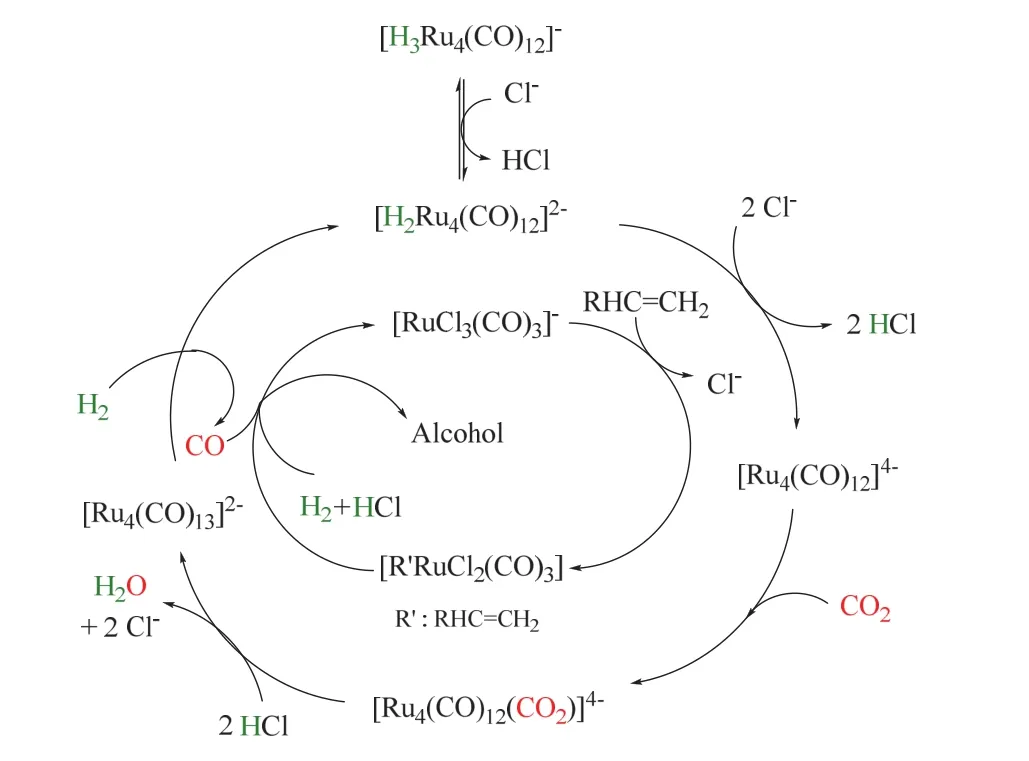
图8 Ru催化CO2/H2参与烯烃羰基化制醇的反应机理Fig. 8 The mechanism of Ru-catalyzed hydroformylation/reduction of alkenes with CO2/H2.
离子液体是指室温或低温下为液体的熔融盐,由有机阳离子和无机/有机阴离子(如卤素离子)组成;离子液体具有极低的蒸气压,这有效消除了挥发性溶剂对环境和人体的危害。基于其各方面优异的特性,离子液体在CO2/H2参与的氢甲酰化反应中可同时充当添加剂和绿色溶剂。Tominaga以1-己烯为底物,在[Bmim]Cl/甲苯或[Bmim][Cl +NTf2](Bmim:1-丁基-3-甲基咪唑;NTf2:二(三氟甲基磺酰)酰亚胺)的混合溶剂中获得了超过80%的庚醇35。Dupont课题组则进一步将离子液体完全替代卤素盐和溶剂的使用,同时H3PO4等酸的引入有利于Ru催化的RWGS,在相对温和的反应条件下,环己烯和多种2,2-二取代烯烃均获得较高的醇收率(如图7b所示)36。
基于上述的研究,Beller课题组考察了多种亚膦酸酯配体,通过具有较大空间位阻的亚膦酸酯配体与Ru3(CO)12相配合,实现了CO2/H2参与的脂肪端烯和内烯烃的氢甲酰化反应,在相对温和的条件下获得醇产物(如图7c所示)。膦配体的引入有效抑制了烯烃的加氢,提高了对应醇的收率37。
羰基钌体系催化RWGS/烯烃氢甲酰化/氢化的串联反应虽然得到了有价值的醇,但催化剂中贵金属钌的用量较高,成本较大,这限制了该类反应的进一步发展。在前述工作的基础上,何林课题组构建了钌钴双金属催化体系(图7d),其中钌基催化体系实现了RWGS/醛氢化,而Co2(CO)8则催化烯烃氢甲酰化,这有效降低了贵金属钌的用量。理论计算表明,体系中酸的引入便于Ru-COOH中间体的脱羟,从而推动了RWGS,使该体系在生成醇的反应中表现出良好的性能。适用烯烃包括多种来自生物质和石油工业的废料,均可通过该策略转化为有价值的醇,这也同时实现了工业废料的增值和CO2利用38。
考虑到钌的稀缺性、高成本以及均相体系中催化剂回收难等问题,发展多相催化体系吸引了很多研究者的兴趣。一方面多相催化剂具备易回收和循环使用的优势,另一方面多相催化剂也在RWGS和传统烯烃氢甲酰化中表现优异,因此,多相催化的CO2/H2参与烯烃氢甲酰化在理论上是可行的。
虽然CO2/H2参与多相氢甲酰化研究的策略与均相类似,均为RWGS和氢甲酰化反应的串联,但依然存在一些挑战。首先是反应温度不匹配:受热力学限制,多相中RWGS所需温度较高(一般高于300 °C),而传统氢甲酰化的温度较低(100 °C左右),实现两个反应的耦合具有一定难度;其次在反应过程中副反应较多:如烯烃加氢、CO2甲烷化等。因此,多相催化体系的创制具有较大挑战性。
Kondratenko课题组在CO2/H2为合成气替代品的多相乙烯氢甲酰化制丙醇的转化中做出了一系列开创性工作。该课题组首次实现Au/TiO2体系催化的CO2/H2参与乙烯氢甲酰化制丙醇的反应39,在2 MPa (CO2/H2/C2H4= 1 : 1 : 1)进料气和200 °C条件下,掺杂K的Au/TiO2催化RWGS生成的CO全部参与到烯烃氢甲酰化反应中,丙醇收率为2.4%,而不掺杂K的Au/TiO2体系丙醇收率仅为0.7%。机理研究发现Au吸附的CO2通过RWGS还原为CO,原位生成的CO 100%选择性的参与乙烯氢甲酰化反应生成丙醛,丙醛进一步加氢生成丙醇。研究表明通过Au沉积方法和K添加剂的引入可以对Au的颗粒尺寸进行调节,且反应的活性和选择性与Au的颗粒尺寸密切相关。K的掺杂不仅能调控Au的尺寸,而且促进吸附氢物种的形成,推动了CH3CH2CO加氢生成丙醇40。然而,K添加剂的掺杂也降低了CO2的转化率,CO2被强吸附在碱性位点上生成离子型碳酸盐(如碳酸钾),而离子型碳酸盐分解为氢甲酰化所需要的CO则非常缓慢。考虑到总体上碱性添加剂K对该反应的积极作用,该课题组考察了碱金属Cs对催化性能的影响41。研究表明Cs与K类似,可明显影响产物的选择性,最终引入Cs的催化体系可获得2.9%的丙醇。实验表征显示Cs存在于载体和Au颗粒之间,这有利于Auδ+的稳定,促进了乙烯的羰基化反应速率,进而提高了丙醇的选择性。
研究发现载体类型对反应性能也有很大影响,该课题组制备了K-Au/TiO2-r (金红石)和Au/TiO2-a (锐钛矿)等催化体系并用于探究不同类型的TiO2载体对反应性能的影响。对比发现KAu/TiO2-r体系更加利于丙醇的生成(详见表1)40,丙醇收率可达3.1%。金属和载体之间的相互作用对金粒径尺寸和分布的影响可能是影响反应的主要原因。该课题组进一步对比了K-Au/TiO2和KAu/SiO2的催化性能(如表1条目2和条目5所示)42,研究发现在TiO2负载的催化剂得到的醇收率比SiO2负载的催化剂更高。实验表征发现TiO2载体表面的碳酸盐和甲酸盐物种不会抑制CO2还原和氢甲酰化反应的发生。反应过程中Auδ+转变为活性位点Au0,乙烯在活性Au0位点被活化,与CO和H2快速反应,最终生成丙醇。而K-Au/SiO2体系更有利于CO2的转化,且随着K含量的提高,SiO2载体促进Au对乙烯和氢气的活化,并显著抑制烯烃加氢。这可能是因为K在SiO2中部分溶解形成硅酸钾层,从而抑制了对RWGS不利的碳酸钾的形成(如前所述,碳酸钾和CO2的结合力更强,使CO2难以被还原为CO) (如图9所示)43。

图9 载体和添加剂对金纳米粒子在CO2/H2和C2H4合成丙醇中的活性和选择性的影响Fig. 9 Effect of support and promoter on activity and selectivity of gold nanoparticles in propanol synthesis from CO2/H2 and C2H4.
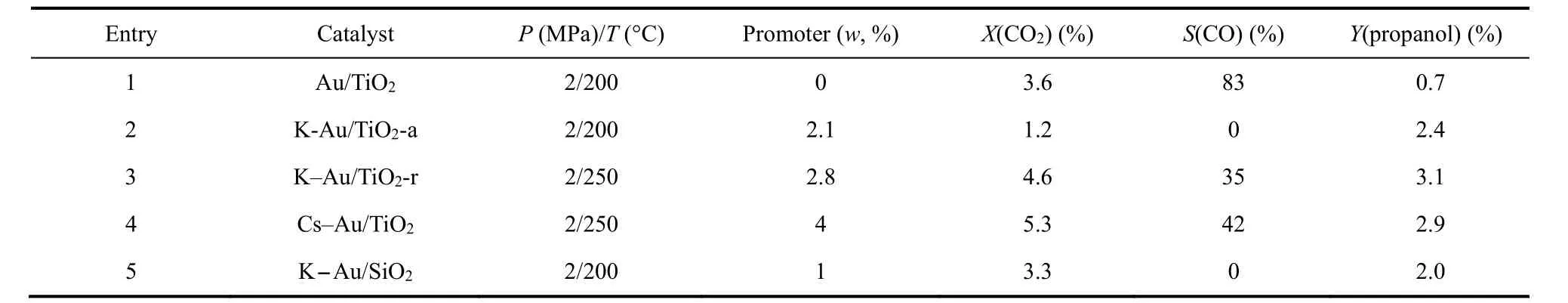
表1 Au基多相催化CO2/H2参与的乙烯氢甲酰化性能比较Table 1 Performance comparison for hydroformylation of ethylene with CO2/H2 catalyzed by Au-based heterogeneous catalysts.
Kondratenko课题组开创了不同于均相羰基钌的Au/TiO2多相催化体系,首次实现了在CO2/H2条件下乙烯到丙醇的转化,阐明了载体和添加剂对催化性能的影响,但在该类催化体系中丙醇的收率较低,反应条件苛刻,后续的探究需进一步提高醇收率和使反应条件温和化。
综上,CO2/H2参与的烯烃氢甲酰化/氢化制醇的反应已经取得了长足的发展,CO2/H2在各种“道具”的帮助下成功突破了众多“关卡”,如以羰基钌为催化剂,添加剂由最初的卤代盐发展到更绿色环保的离子液体。在温和反应条件下,实现了多种环烯烃、2,2-二取代烯烃等的高效转化成醇,膦配体与羰基钌的成功结合也进一步实现了脂肪端烯和内烯烃的高效转化,同时钌钴双金属体系的开发降低了贵金属的使用。值得关注的是,Au/TiO2多相催化体系的开发成功实现了乙烯到丙醇的转化,拓宽了该类反应的催化剂类型,为催化剂的循环再利用提供了基础。虽然通过离子液体和配体分别促进了脂肪端烯的醇收率和区域选择性的一定提升,但获得高化学/区域选择性的醇/醛这一有挑战性的“关卡”仍需进一步探索。
2.2 CO2/H2参与的烯烃及其衍生物的氢羧基化制高级羧酸
在CO2作为羧基源的烯烃还原羧化生成高级羧酸的研究中,如图10a所示,还原剂主要使用超化学计量的乙基锌、镁试剂、金属锰等有机金属试剂或有机硅试剂44-47,这些试剂一般来说价格昂贵,且对空气/水敏感,这限制了该催化方法的应用。H2是一种清洁、廉价易得的还原剂,基于CO2/H2的烯烃还原羧化制高级羧酸是更具前景和挑战性的(图10b)。
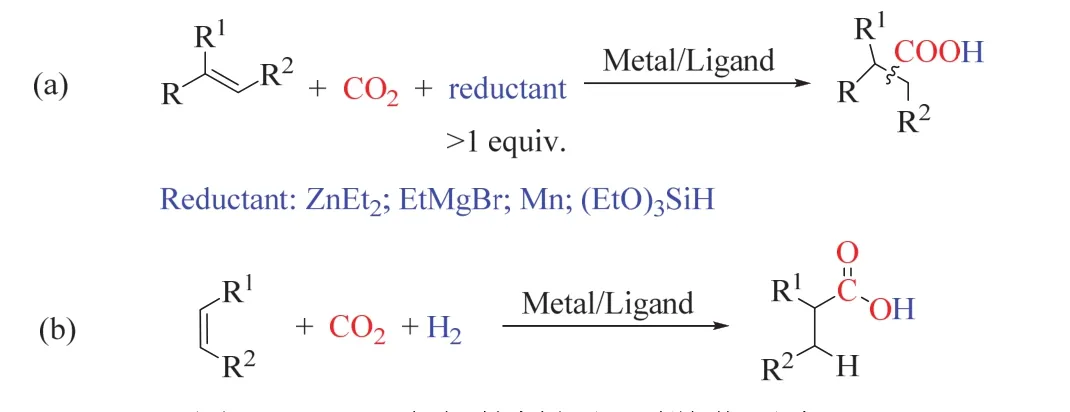
图10 CO2参与的烯烃还原羧化反应Fig. 10 Reduction carboxylation of alkene with CO2。
Leitner课题组提出了从CO2、H2和烯烃直接合成高级羧酸的催化策略(图11)48。该策略以[RhCl(CO)2]2为金属前体,PPh3为配体,碘甲烷(CH3I)和对甲苯磺酸(PTSA)的引入促进了氢羧化反应的进行,多种烯烃在180 °C、CO2/H2(60/10 bar,1 bar = 0.1 MPa)的条件下反应生成增一个碳的羧酸,其收率最高可达92%。实验研究显示单齿膦配体通常适合用于该反应体系,而多齿膦配体在该反应中则完全抑制了羧酸的形成,可能的原因是多齿膦配体抑制了RWGS的进行。对反应历程的探究表明,乙酸环己酯可能为反应活性最高的中间物,醇也因此可充当有效的底物,其转化率和产物分布与对应的烯烃基本一致。同位素实验表明,羧酸基团并非由CO2分子直接嵌入构筑,而是经历了CO和H2O作为活性物种的传统氢羧化途径。整个转变可能通过Rh催化的RWGS反应、原位生成的CO和H2O随后参与烯烃的氢羧化反应。该反应把逆水煤气变换中的产物H2O也利用起来,实现了100%的原子利用率。
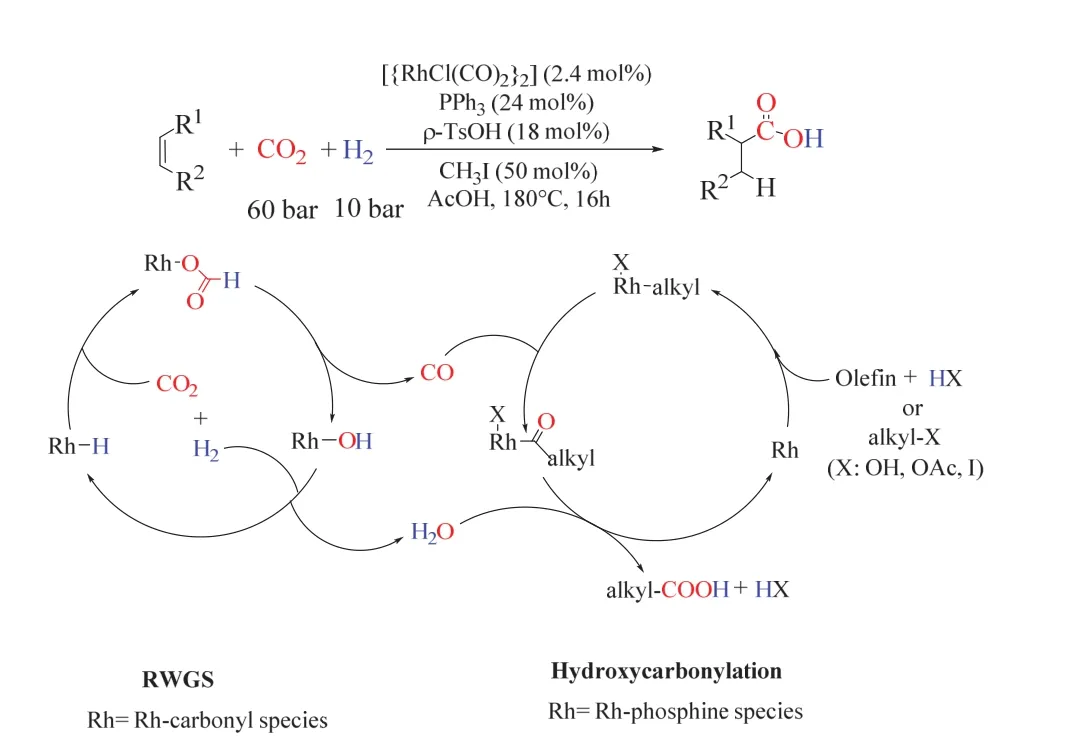
图11 CO2为C1源的烯烃氢羧化反应生成高级羧酸Fig. 11 CO2 as a C1 building block for the formation of carboxylic acids by hydrocarboxylation.
该策略提供了一种新的以CO2为C1源合成高级羧酸的方法,实现了从CO2/H2组合出发制备高级羧酸的“关卡”的突破。近期韩布兴课题组报道的Ir催化CO2/H2参与的醚制高级羧酸。在该催化体系下醚会首先转化为烯烃,随后与原位生成的CO发生羰基化反应。因此,我们将醚视做烯烃的衍生物。
一般来说,醚的反应活性低于烯烃或醇,且没有作为底物和CO2/H2制备高级羧酸的前例。韩布兴课题组首次实现了由醚、CO2和H2合成高级羧酸,在170 °C、5/2 MPa CO2/ H2氛围下,以LiI为促进剂的IrI4体系高效催化该转化(如图12所示),获得了优异收率的高级羧酸49。该催化体系具有良好的底物普适性,各种醚如环醚、直链烷基醚、支链烷基醚和芳香醚等都能被高效转化为相应的高级羧酸。机理研究表明,底物醚被快速选择性地转化为烯烃,这是该策略顺利进行的关键步骤,烯烃与I-进一步转化为烷基碘化物,RWGS原位产生的CO插入到烷基碘化物中,随后被原位形成的水还原消除,最终生成增一个碳的羧酸。该方法中底物醚相对烯烃更为廉价易得,具有较大的应用前景。
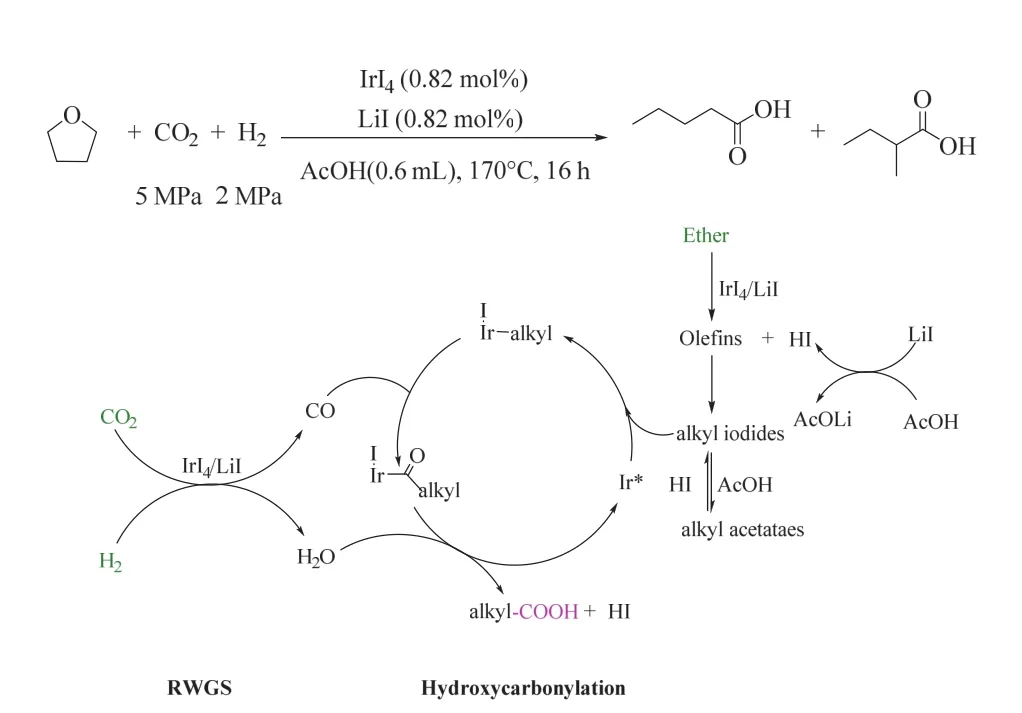
图12 IrI4催化CO2/H2参与的醚羰基化生成羧酸的反应49Fig. 12 Ir-catalyzed synthesis of carboxylic acid from ether, CO2/H2 49.
综上,在CO2/H2参与烯烃及其衍生物醚的氢羧化生成高级羧酸的反应过程中,Rh和Ir均成功实现了RWGS和烯烃氢羧化反应的高效耦合,得到了增一个碳的高级羧酸。该类反应实现了原子的高效利用,但催化性能还有待提升,如需要相对苛刻的反应条件以及较大剂量的添加剂。因此,反应条件温和化,并减少添加剂的使用等是下一步研究重点。
2.3 CO2/ROH参与的烯烃氢酯化制酯
烯烃羰基化反应不仅可提供醛、醇,还可提供工业中重要的聚合物中间体羧酸酯(这类转化也称为氢酯化反应),如目前工业上生产聚甲基丙烯酸甲酯(有机玻璃)的工艺便是基于钯催化的乙烯氢酯化反应50-52。
Beller课题组发展了一种新颖的烯烃氢酯化反应53, 以CO2作C1源,MeOH为还原剂,避免使用敏感或昂贵的还原剂。该催化体系以Ru3(CO)12为催化剂,离子液体[Bmim]Cl为添加剂,在相对温和的条件下实现了烯烃氢酯化反应,最终得到产物酯。脂肪烯烃和芳香烯烃在该催化体系下均可高效转化为对应的羧酸酯,如从乙烯出发生成工业中重要的丙酸甲酯;此外,乙醇和苯甲醇替代甲醇在该体系中也能实现很好的转化。作者通过同位素标记和控制实验对反应机理进行了探究,发现CO2是主要的羰基来源,同时CH3OH也可充当羰基源,基于此,作者提出了几种可能的途径,分别经历CO和甲酸甲酯中间体(图13)。该策略首次实现了从CO2/醇出发合成羧酸酯,这种转化进一步拓宽了CO2作为C1源参与形成C―C键的催化领域。

图13 钌催化CO2/醇参与烯烃氢酯化反应53Fig. 13 Ru-catalysed alkoxycarbonylation of alkenes with CO2/alcohols 53.
上述氢酯化过程包含CO2还原和烯烃氢酯化两个步骤,使用单一催化剂同时高效催化该串联反应是困难的,这是因为各反应对催化剂的要求是不同的。基于这一考虑,何林课题组发展了钌-钴双金属协同催化CO2还原/烯烃氢酯化的体系(图14)54。该双金属体系表现出良好的催化性能和可重复使用性,与之前单钌的催化体系相比,二元催化体系可有效地减少贵金属和离子液体添加剂的使用。机理研究表明钌催化剂主要参与CO2还原,环己烯的氢酯化则依赖于钴催化剂。密度泛函理论(DFT)计算从能量的角度阐明了[Ru(CO)3Cl2]2缓慢分解形成活性钌催化剂以及烯烃氢化的发生。
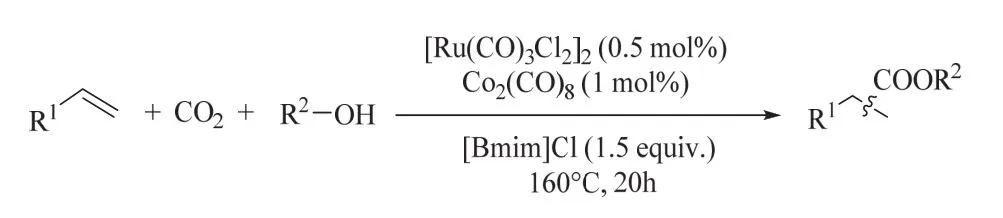
图14 钌-钴双金属协同催化CO2/醇参与烯烃氢酯化Fig. 14 Alkoxycarbonylation of alkenes with CO2/alcohols by a reusable heterobimetallic ruthenium cobalt catalytic system.
羰基钌催化的CO2/ROH参与的烯烃氢酯化反应开辟了新的合成路线,从烯烃、CO2/醇得到了之前未能实现的酯的产物,但该体系钌用量较高,反应条件较苛刻。因此,开发更加温和、高效的催化体系仍是令人期待的。
2.4 CO2/H2参与的烯烃氢甲酰化/还原胺化制胺
与其他烯烃官能团化相比,胺的合成在有机化学中也引起越来越多的关注55。除了作为大宗化学品,由于其优异的生物活性,仲胺和叔胺也可作药物发现方面重要的靶标。本节基于前述Ru催化CO2/H2参与烯烃氢甲酰化,研究者向该体系中引入伯胺或仲胺,原位生成的醛与胺还原胺化,分别生成仲胺或叔胺。值得一提的是,本文主要涉及经典的C―C键构筑的羰基化反应,而如CO2/H2参与的胺羰基化/胺甲基化等直接形成C―N键的羰基化反应也得到广泛报道23,56,但不在本节讨论范围内。
Eilbracht等报道了首例Ru催化的RWGS/烯烃氢甲酰化/还原胺化多步串联反应,该催化体系以Ru3(CO)12为催化剂,LiCl为添加剂,苄基三乙基氯化铵(BTAC)为相转移剂,在CO2/H2(20/60 bar),160 °C条件下反应5天后,能以35%-98%的收率得到多种期望的仲或叔胺(图15)57。该体系顺利进行的关键在于LiCl作为添加剂抑制烷烃、醇、甲酰胺等副产物的形成,相转移催化剂的加入提高了LiCl的溶解度,并对整体选择性产生了积极影响。在该策略下,醛加氢到醇的过程被抑制,醛被胺捕获发生不可逆地缩合并还原,最终得到胺的产物。

图15 钌催化CO2/H2和胺参与的烯烃氢胺甲基化Fig. 15 Ru-catalyzed hydroaminomethylation of alkenes with CO2/H2 and amines.
在上述工作的基础上,Dupont课题组将Ru3(CO)12溶解在离子液体中,同样高效的实现了该串联转化:(a) RWGS,(b) 烯烃氢甲酰化,(c)醛的还原胺化。在120 °C,CO2/H2(60 bar,1 : 1)条件下,烯烃与伯胺或仲胺(烯烃/胺= 1 : 1)在Ru3(CO)12催化下反应,反应时间缩短至36 h,烯烃转化率高达99%,胺的选择性高达96%58。该体系比之前报道的体系催化效率更高,同时降低了贵金属的使用量,并将反应条件进一步温和化。
尽管反应路径复杂,CO2/H2参与的烯烃氢甲酰化/还原胺化仍实现了仲胺或叔胺的高效制备,后续可在更具普适性的烯烃和胺底物、更趋温和的反应条件以及更短的反应时间等上实现该“关卡”的突破。
2.5 CO2/H2参与的烯烃氢甲酰化制醛
如前所述,CO2/H2参与烯烃羰基化可获得醇、羧酸、酯、胺等产物,其反应策略主要基于过渡金属催化RWGS得到CO的反应路径,但RWGS所需的高温和钌的过度氢化能力使烯烃氢甲酰化中目标产物不能停留在醛,而是进一步氢化生成醇。而中间体醛作为大宗化学品在工业中具有非常重要的地位。为了选择性得到醛的产物,丁奎岭课题组经过巧妙的策略设计,以Rh(acac)(CO)2和螺环骨架的亚膦酸酯配体为催化剂,并加入额外的还原剂PMHS,最终实现了温和条件下(100 °C,5/20 bar CO2/H2)的烯烃氢甲酰化反应,获得高化学/区域选择性的直链醛,其中醛收率可达70%,直异比可达10 : 1 (图16)59。作者通过控制实验证实了CO的生成来源于CO2与PMHS反应,而非RWGS,这避免了RWGS所需的苛刻条件,同时温和的条件有利于提高醛的直异比。CO2被PMHS还原优先于醛的还原以及Rh/亚磷酸酯催化体系的弱氢化能力是反应得以停留在醛的关键。
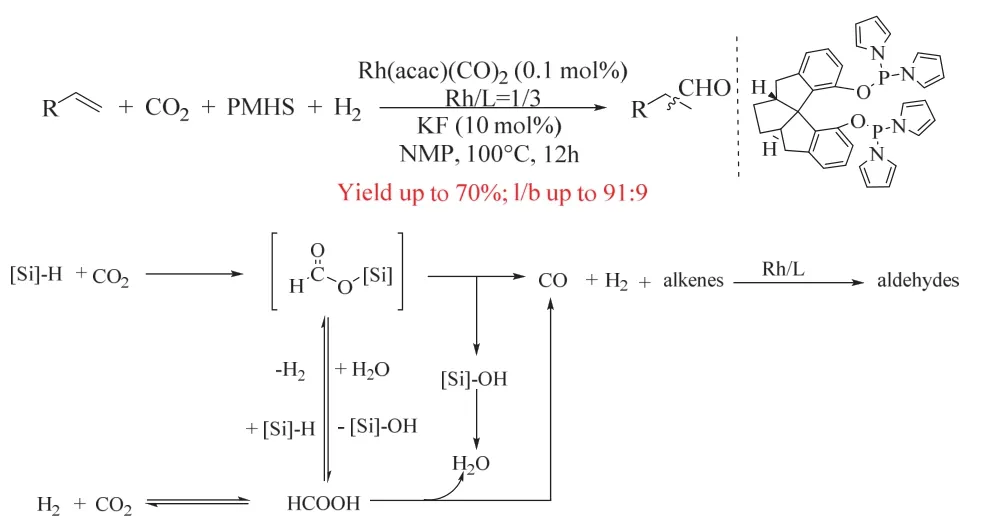
图16 铑催化CO2和氢硅烷参与的烯烃氢甲酰化反应Fig. 16 Rhodium complex-catalyzed hydroformylation of alkenes with CO2 and hydrosilane.
通过引入额外的还原剂PMHS,CO2/H2参与的烯烃氢甲酰化可选择性获得直链醛,这为CO2/H2参与的烯烃羰基化反应提供了一种新的思路。然而,额外还原剂PMHS的使用造成了部分底物的损失和产物分离的困难。因此,以H2为唯一还原剂的CO2/H2作合成气替代品的烯烃氢甲酰化反应仍是令人期待的。
最近,陈经广课题组通过串联的固定床成功将CO2、乙烷脱氢重整和氢甲酰化高效耦合,实现了丙醛和丙醇的生成60。该策略采用多相Fe3Ni1/CeO2体系催化CO2辅助乙烷的脱氢重整,获得乙烯、CO和H2;而RhCox/MCM-41催化剂则顺利促进乙烯的氢甲酰化反应选择性生成C3含氧化物(丙醛和丙醇) (如图17所示)。该策略在无外加还原剂的存在下实现了CO2/C2H6两种惰性、廉价分子转化为高价值化学品醛和醇,为CO2参与的羰基化反应提供了新的思路。
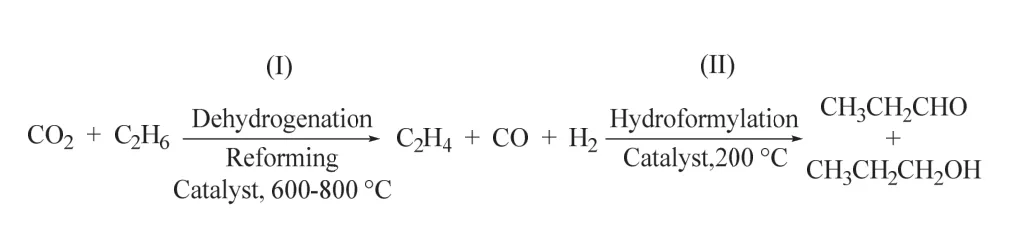
图17 多相催化CO2和乙烷反应制丙醛/丙醇Fig. 17 The conversion of CO2 and ethane to propionaldehyde/propanol catalyzed by heterogeneous catalyst.
3 CO2/H2参与的卤代烃羰基化反应
芳香醛是C―C、C―N和C―S偶联反应中的重要中间体,在材料、药品、农药和农用化学品的合成中有着广泛的应用61。合成芳香醛的传统方法包括羧酸或酯还原,Gattermann-Koch、Reimer-Tiemann、Duff和Vilsmeier反应等62,这些方法通常存在某些固有缺陷,如收率低、选择性差、需要有毒气体和苛刻条件等。基于此,利用CO或CO替代品作为C1源,从芳香卤化物直接甲酰化合成芳香醛的转化逐渐发展起来。如图18a-c所示,CO或甲醛,甲酸等CO替代品均能在钯催化下高效生成芳香醛63-65。这一进展也促进了使用CO2代替CO进行芳香卤化物甲酰化的探索。刘志敏课题组在CO2参与的芳香卤代物羰基化反应中做出了开创性的工作,首次以CO2为C1源,在氢硅烷PMHS和碱1,8-二氮杂二环十一碳-7-烯(DBU)的存在下,实现了Pd催化芳香碘/溴化物66,67直接甲酰化生成芳香醛(图18d)。

图18 钯催化CO及其替代品的芳基卤代烃甲酰化反应Fig. 18 Palladium-catalyzed formylation of aryl halides with CO and its derivatives.
在上述工作的基础上,同一课题组首次使用CO2/H2参与芳香卤化物的甲酰化转化(图19)68。该催化体系由RhI3(适用于芳香碘化物)或RhI3/Pd(dppp)Cl2(适用于芳香溴化物)和PPh3组成,乙酸酐(Ac2O)为脱氧试剂,并以三乙胺(Et3N)为碱性添加剂,该体系在100 °C下以优异的收率得到芳香醛,并表现出良好的官能团耐受性和广泛的底物适用性。控制实验表明反应经历了三个串联步骤,分别是在Et3N辅助下CO2加氢生成HCOOH(或HCOOH-NEt3加合物),其在Ac2O存在下分解、释放CO,原位生成的CO与芳香卤化物经历传统的甲酰化反应生成芳香醛。
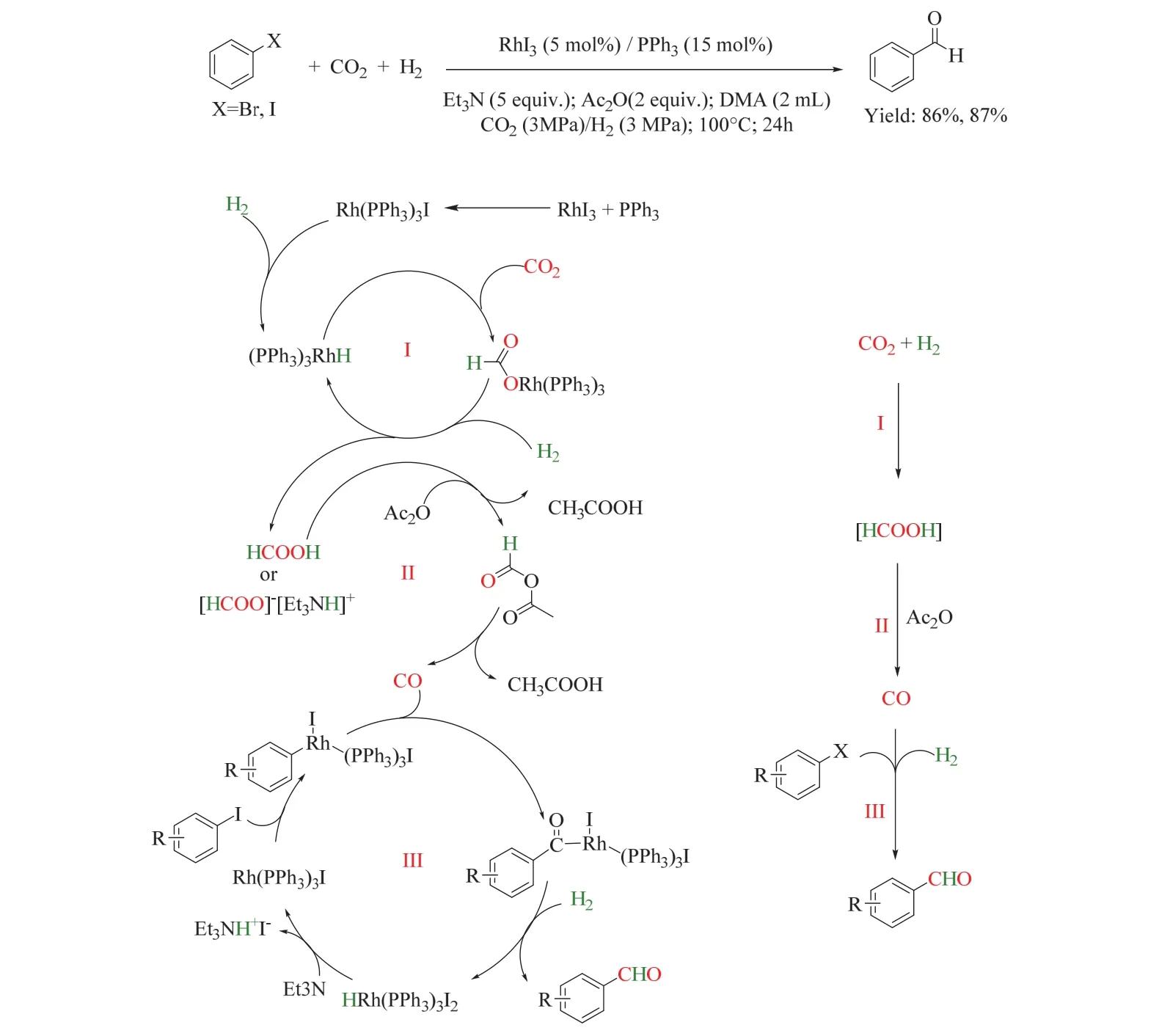
图19 铑催化的CO2/H2参与的芳香卤代烃甲酰化Fig. 19 Rhodium-catalyzed formylation of aryl halides with CO2 and H2.
这种不同于RWGS路径的、经历甲酸中间体的多步串联的反应路径实现了温和条件下CO2氢化间接转化为CO,为CO2/H2实现更多的羰基化反应提供了兼容的可能性。
4 CO2/H2参与的甲醇及其衍生物的羰基化反应
乙酸是一类重要的基础大宗化学品,可用作合成乙酸乙烯酯单体和乙酸酐的原料,目前工业中主要通过甲醇羰基化的方法生产,以来自于化石燃料的CO为羰基源69-71。利用甲醇及其衍生物(如甲氧基醚等富含OCH3的化合物)和CO2/H2合成乙酸等化学品对绿色可持续发展具有重要意义,但也具有很大挑战性72,73。
乙醇作为一种重要的大宗化学品,被广泛用作溶剂、食品、医药、农药、有机原料等,同时作为可替代燃料在当前的基础能源设施中发挥着日益重要的作用。目前,乙醇主要是通过石油生成的乙烯催化水合或玉米、甘蔗等食物发酵生产。因此,以CO2和H2为原料合成乙醇具有重要意义,但这仍然是一个巨大的挑战,因为它涉及到CO2加氢的同时形成C―C键73,74。使用CO2合成乙醇的报道大多局限于高温下CO2加氢,该路线中,CO2通常与H2反应生成高活性C1中间体,包括CO、CH3*(吸附态的CH3)或CH3OH,这些中间体经过C―C键偶联形成C2+产物,例如乙醇和高级醇75,76。由于CO2氢化原位生成C1中间体和碳链增长同时发生,因此产物分布通常较宽,乙醇在醇类产物中的占比较低。为了提高乙醇的选择性,引入特定底物与CO2和H2反应是一种可行的方法。例如以甲醇、CO2和H2为起始原料,乙醇是唯一醇类产物,其他甲醇衍生物如甲醛、二甲醚(DME)等,也可作为底物与CO2/H2反应选择性生成乙醇。
本节总结了近年来CO2/H2参与的甲醇或其衍生物羰基化制乙酸、乙醇的报道,其中韩布兴课题组在该领域做出了开创性的工作。
4.1 CO2/H2参与的甲醇及其衍生物羰基化制乙酸的反应
利用可再生的、廉价的CO2合成乙酸具有重要意义,但同时也具有很大挑战性。目前CO2参与乙酸合成的路线存在明显的局限,如选择性低、催化剂活性低、反应温度高、使用昂贵或有毒的反应物等。例如,以CO2和CH4为原料合成乙酸受限于热力学限制,往往需要很高的温度,且乙酸的产率较低77。而使用CH3I、CO2和H2为反应物时,可获得选择性10.7%的乙酸,但CH3I高毒且价格昂贵。因此,实现CO2高效、高选择性生成乙酸是令人期待的78。
韩布兴课题组报道了甲醇、CO2和H2合成乙酸的新型反应路径(图20)79。该催化体系以Ru3(CO)12/Rh2(OAc)4为催化剂,咪唑为配体,LiI为促进剂,在180 °C以上可高收率的获得乙酸,其转化频率(TOF)值达到30.8 h-1,基于甲醇的乙酸收率高达77%,且催化剂经过5次循环后,催化性能几乎保持不变。体系中配体咪唑对催化剂的高催化活性、稳定性和选择性起着关键作用。令人意外的是,乙酸生成的反应不通过CO参与的反应路径,CO在反应的气体样品中几乎检测不到。随后,作者通过同位素实验证实CO2是通过直接插入参与的羰基化反应。反应机理如图所示:甲醇与LiI首先形成CH3I (步骤1),随后CH3I与活性Rh*生成CH3Rh*I (步骤2),CO2直接插入CH3Rh*I中形成CH3COORh*I (步骤3),该过程中Rh催化乙酸的生成。在H2的存在下活性Ru物种(Ru*)促进CH3COORh*I还原消除生成乙酸(步骤4),最后,原位生成的LiOH和HI中和形成LiI和H2O (步骤5)。所有的催化物种再生后进行下一个循环。
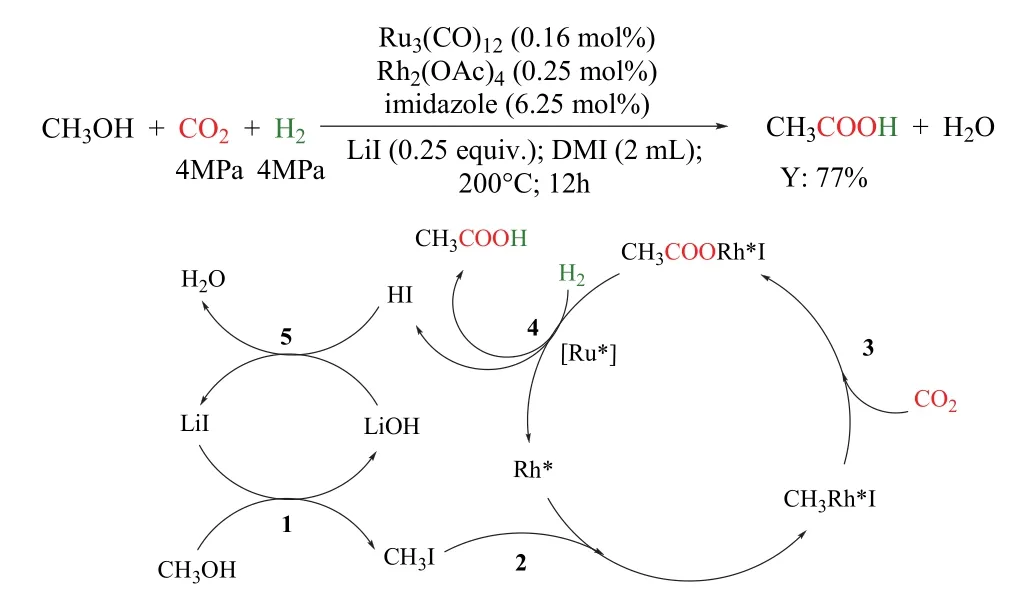
图20 Ru-Rh双金属催化的CO2/H2参与的甲醇羰基化制乙酸79Fig. 20 Synthesis of acetic acid via methanol hydrocarboxylation with CO2 and H2 by Ru-Rh bimetallic catalyst 79.
在此工作的基础上,同一课题组将双金属体系发展为单金属Rh2(CO)4Cl2/4-甲基咪唑(4-MI)/LiI体系(如图21所示)。该反应在150 °C时乙酸开始形成,在180 °C时乙酸收率最高可达81.8%,此时TOF高达26.2 h-1。与双金属体系相比,该催化体系更简单,且在温和的条件下效率更高80。
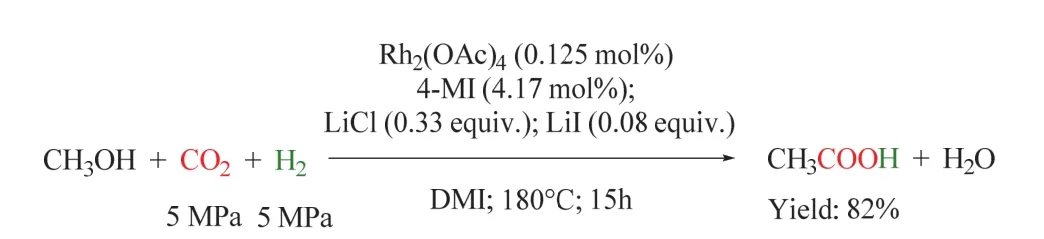
图21 Rh催化的CO2/H2参与的甲醇制羧酸反应Fig. 21 Synthesis of carboxylic acid from methanol with CO2/H2 by Rh catalyst.
众所周知,木质素是一类拥有丰富含氧基团(例如OCH3)的生物高聚物,其中的OCH3可充当甲醇衍生物,参与到羰基化反应中。基于此,刘志敏课题组使用前述类似的双金属催化体系,实现了苯甲醚(木质素中最简单的平台化合物)在CO2/H2条件下合成乙酸,其中苯甲醚可提供甲基生成CH3I。在180 °C,3/3 MPa CO2/H2条件下时,乙酸的收率可达94% (图22),值得注意的是,离子液体(如[Bmim]Cl)充当溶剂并促进了催化活性中心的形成,机理探究表明[BMIm]+稳定的RhI3和I-络合的Ru(CO)n(n = 1-4)是形成乙酸的关键活性催化物81。其后,韩布兴课题组以离子液体1-己基-3-甲基咪唑四氟硼酸盐(HMimBF4)为添加剂,在相对温和的条件下也实现了苯甲醚/木质素与CO2、H2生成乙酸的转化82。
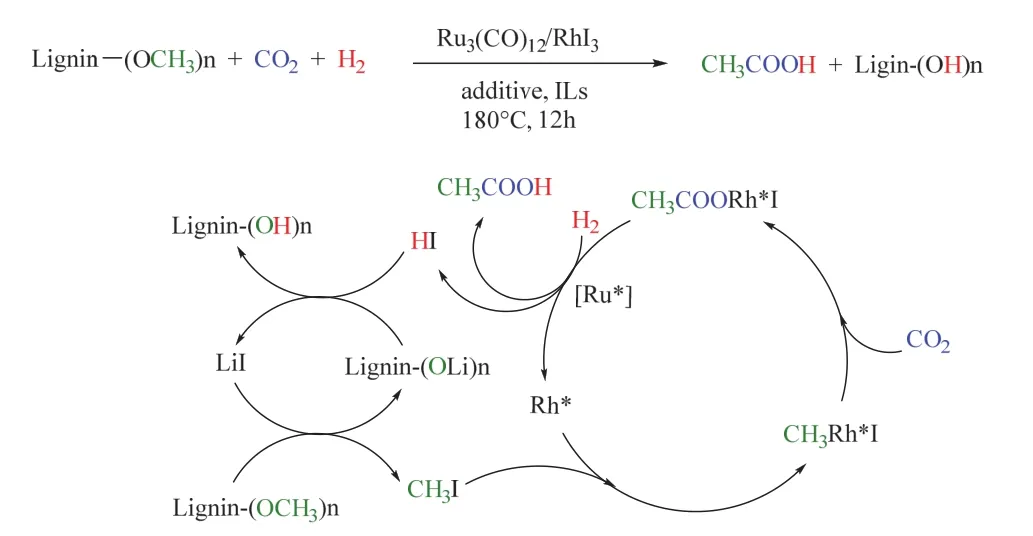
图22 Ru-Rh双金属催化CO2/H2参与木质素羰基化制乙酸Fig. 22 Ru-Rh bimetal-catalyzed carbonylation of lignin to acetic acid with CO2/H2.
综上,CO2/H2参与甲醇及其衍生物羰基化制乙酸的反应近年来得到快速发展,Rh/Ru双金属体系首次实现CO2直接插入的羰基化反应生成乙酸,揭示了CO2/H2参与羰基化反应的新的反应路径。在保证乙酸选择性和收率的基础上,通过配体筛选(咪唑到4-甲基咪唑),添加剂(卤素盐到离子液体)和反应条件的优化,实现了单金属Rh高效催化该反应,提高了催化性能,降低了贵金属的使用,同时实现了苯甲醚/木质素等富含OCH3的甲醇衍生物与CO2、H2生成乙酸的转化,拓宽了该类反应的底物范围。但该体系铑用量较高,反应条件较苛刻。后续可在更温和的反应条件以及更多含OCH2-R类型的化合物(如甲醛、乙醇等)上实现多种羧酸“关卡”的突破。
4.2 CO2/H2参与甲醇及其衍生物羰基化制乙醇的反应
在多数情况下,C2+产品,如乙醇、乙酸、烯烃和液体燃料等相对C1产品用途更为广泛。但CO2和H2高效、高选择性地直接合成C2+产物相对困难,因为这同时涉及到可控的CO2加氢和C―C键形成,这在基础科学中仍然是一个挑战73,83-86。目前,CO2加氢直接制乙醇的反应温度普遍较高,反应活性和选择性较低。基于此,引入特定底物如甲醇或其衍生物(如甲醛、二甲醚等)与CO2/H2反应合成乙醇是提高催化活性和选择性的可行方法。从反应机理的角度考虑,反应一般被认为经历了源自RWGS的CO插入生成乙醛的过程,因此可被归到羰基化反应之列。
1998年Sasaki课题组首次报道了CO2/H2参与的甲醇同系化制乙醇的反应(如图23所示),该体系以Ru-Co双金属为催化剂,两种金属间存在协同作用,乙醇的收率受所加添加剂卤代盐的显著影响,其中LiI性能最佳。10 mmol甲醇在180 °C和2/10 MPa CO2/H2条件下反应15 h,乙醇收率最高可达3.2 mmol,乙醇选择性为34.2 C-mol% (代表以碳摩尔数计的产率)87。作者推测甲醇被来自于CO2的CO所同系化生成乙醇。尽管该体系的催化性能有待提高,但它为高选择性制乙醇开辟了新的途径。
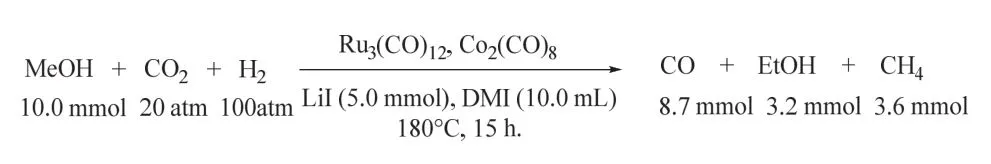
图23 钌-钴双金属催化CO2/H2参与甲醇体系化的反应Fig. 23 Methanol homologation using CO2/H2 catalyzed by ruthenium-cobalt bimetallic complex system.
基于上述工作,韩布兴课题组在2017年报道了多聚甲醛(可视作甲醇的衍生物)为反应底物、CO2和H2参与合成乙醇的反应88。该体系以LiI作为促进剂,在大于140 °C,CO2/H2(3/5 MPa)条件下,Ru-Co双金属催化剂可高效协同催化该转化,基于金属Ru的乙醇TOF值最高可达17.9 h-1,乙醇在总产物中的选择性达到50.9 C-mol% (如图24所示)。此外,该催化体系能多次重复使用,在5个循环后乙醇的总转化数(TON)超过805。机理研究表明该转化由三个串联反应组成,分别是:甲醛氢化生成甲醇,RWGS得到CO,甲醇被CO同系化,最终生成乙醇。基于对机理的深入探究,同一课题组把引入的特定底物从甲醛拓展到二甲醚89、甲醇90,91和芳基甲基醚/木质素92,在相似的催化体系下同样顺利实现了CO2/H2参与的乙醇合成。其中以甲醇为特定底物时,实现了单一金属Ru促进的乙醇生成90,且使用离子液体作为溶剂,扩展了这类反应的溶剂范围。
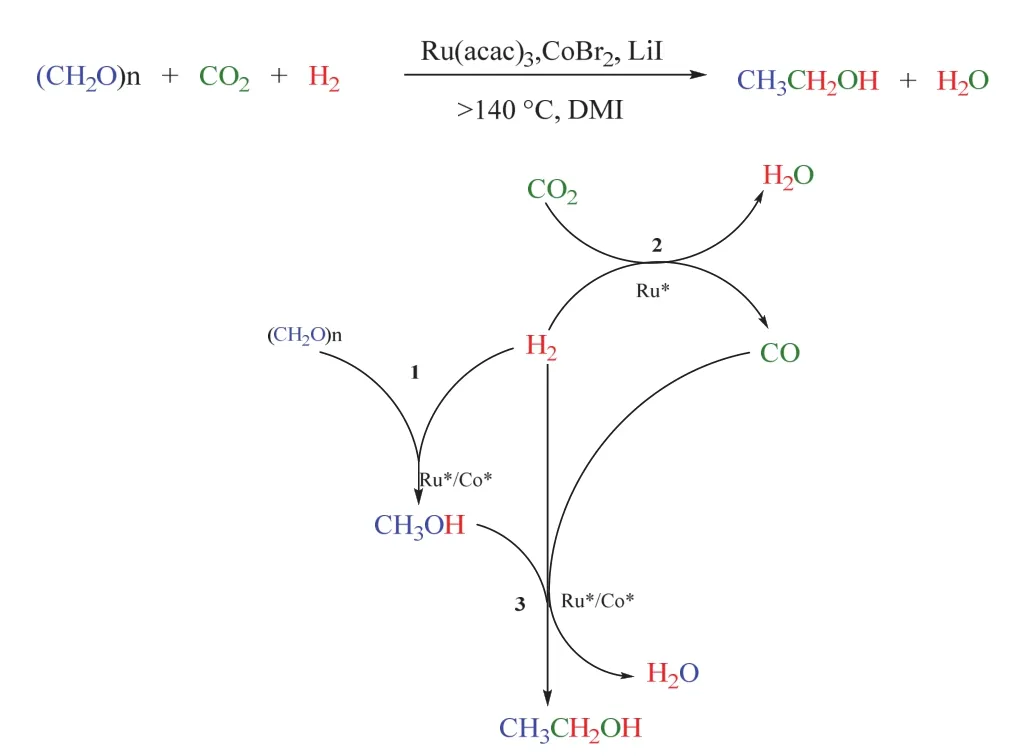
图24 Ru-Co催化的多聚甲醛、CO2和H2合成乙醇Fig. 24 Ru-Co catalyzed synthesis of ethanol from paraformaldehyde, CO2 and H2.
综上,CO2/H2参与甲醇及其衍生物羰基化制乙醇的反应得到进一步发展,利用可多次重复使用的Ru-Co双金属高效协同催化该转化,实现了RWGS/甲醇同系化制乙醇,通过添加剂筛选和反应条件的优化进一步提高了乙醇的选择性,也实现了单一金属Ru催化的乙醇生成。同时该类转化的底物也由甲醇拓展到甲醛、二甲醚和芳基甲基醚等,拓宽了该类转化的底物范围。后续可在更温和的反应条件以及更多含OCH2-R类型的化合物上实现多元醇“关卡”的突破。
5 结论与展望
CO2作为一种丰富、廉价的C1源被广泛用于生产多种高价值的化学品,H2作为最有前景的还原剂之一,与CO2的组合在过渡金属的催化下实现了多种羰基化反应,可获得诸如醛、醇、羧酸、酯、胺等高附加值化学品。在这类反应中,过渡金属(钌、铑、铱)催化的RWGS生成CO是此类转化的关键步骤,随后原位生成的CO随后参与到羰基化反应中。通过向催化体系中引入卤代盐或离子液体、膦配体、非贵金属钴等手段一定程度上抑制了烯烃氢化、提高了区域选择性和反应效率,降低了贵金属用量。同时多相Au基催化剂的开发为催化剂设计提供了一条新的思路。此外,甲醇及其衍生物的羰基化高化学选择性、高收率的生成乙醇进一步拓展了CO2/H2参与的羰基化反应的领域。除了需高温高压的RWGS反应路径,CO2加氢生成甲酸、随后分解为CO的反应路径也被初步探索,实现了温和条件下CO2间接转化为CO,并与芳香卤代物的甲酰化高效耦合,实现了CO2/H2参与构筑了甲酰基。与CO2氢化为CO参与羰基化的反应路径相比,CO2直接参与羰基化的反应路径丰富了CO2/H2参与羰基化的模式,实现了CO2作为羰基源的甲醇制乙酸的反应。这些CO2/H2参与的不同类型的羰基化反应和模式拓展了CO2资源化利用的途径,获得高附加值的大宗或精细化学品,促进了绿色化学的发展。
尽管CO2/H2参与的羰基化反应已经有较多且深入的研究,但这些催化体系距离高化学选择性、高区域选择性以及潜在的工业化还有很长一段路要走,主要原因如下:
(1) RWGS路径和CO2直接插入的反应路径常需高温高压,反应条件苛刻,这限制了配体发挥作用,导致了较低的催化性能和化学/区域选择性,如烯烃氢甲酰化/氢化所得到的醇直异比整体都较差。而CO2加氢到甲酸、甲酸分解为CO的过程中需要消耗(超)化学计量的乙酸酐,这些均限制了CO2/H2参与羰基化反应的进一步发展。
(2) 绝大多数CO2/H2参与的羰基化反应常需要含量较高的添加剂以抑制副反应的发生。
(3) 贵金属用量过高,催化剂的高效回收再利用需要整体解决方案。
基于上述存在的问题,开发新颖的催化体系和羰基化反应类型仍是重要的研究方向。为了实现这些“关卡”,完成这些挑战,以下几个方面值得进一步深入探究:
(1) 实现温和条件下的RWGS,并与后续的过渡金属/配体催化的传统羰基化相耦合,实现高化学/区域选择性的羰基化。
(2) 实现CO2加氢到甲酸、甲酸无需当量添加剂的消耗即分解为CO、CO参与羰基化反应的高效串联耦合。有研究表明,甲酸可在含有内置碱的配体作用下选择性分解成CO,这为该多步串联反应的设想提供了可行性。
(3) 减少该类反应贵金属的使用量,提高金属催化剂的催化性能,也可通过非贵金属部分或全部替代贵金属来催化CO2/H2参与的羰基化反应。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Understanding the Role of Cu/ZnO lnteraction in CO2 Hydrogenation to Methanol
- Electrocatalytic CO2 Reduction to Ethylene over CeO2-Supported Cu Nanoparticles: Effect of Exposed Facets of CeO2
- Controlling the Global Mean Temperature by Decarbonization
- Cu@UiO-66 Derived Cu+-ZrO2 Interfacial Sites for Efficient CO2 Hydrogenation to Methanol
- 二氧化碳电还原反应的理论研究
- 离子液体介导CO2化学转化研究进展