Effects of sepsis and its treatment measures on intestinal flora structure in critical care patients
2021-05-25XiaoJuanYangDanLiuHongYanRenXiaoYaZhangJunZhangXiaoJunYang
Xiao-Juan Yang, Dan Liu, Hong-Yan Ren, Xiao-Ya Zhang, Jun Zhang, Xiao-Jun Yang
Abstract
Key Words: Sepsis; Intestinal flora; 1 6 S rRNA gene sequencing; Dynamic changes;Intestinal barrier index; Procalcitonin
INTRODUCTION
Sepsis refers to the life-threatening organ dysfunction caused by imbalanced responses to infection[1]. As the largest reservoir of bacteria and endotoxins in the body, the intestinal tract is regarded as the “engine” of sepsis and multiple organ dysfunction syndrome. Under normal circumstances, the intestinal flora plays beneficial roles in the context of human physiology and immunity[2 -3 ], existing in a homeostatic state important for the maintenance of human health. However, in the context of critical illness, homeostasis is interrupted, leading to abnormal changes in the types,quantities, proportions, and locations of microorganisms in the intestinal tract[4], thus increasing susceptibility to sepsis. In fact, studies have shown that there is a dose–response relationship between the degree of intestinal microecological disturbance and the incidence of subsequent severe sepsis[5]. Therefore, an improved understanding of the status and degree of intestinal flora disturbances in sepsis patients is of great significance; for instance, the establishment of strong intestinal microecology-based biomarkers will allow for the accurate prediction of prognosis of sepsis patients and the consequent adoption of adequate treatment measures.However, there are few reports on the sepsis-related intestinal flora and its dynamic changes.
High-throughput16S rRNAgene sequencing allows for a comprehensive understanding of the intestinal flora structure, and it is the standard technology used for human intestinal flora analysis[6]. In this study, the diversity and composition of the intestinal flora in sepsis patients, non-sepsis patients, and healthy individuals were analyzed and compared. The dynamic changes in the intestinal flora α-diversity and structure throughout a 1 wk period [1 , 3 , and 7 d after admittance to the intensive care unit (ICU)] were also investigated to disclose the transformation of the intestinal flora in the context of antibiotic treatment. In addition, the potential relationships between the intestinal flora imbalance and clinical indicators in sepsis were investigatedviacorrelation analysis.
MATERIALS AND METHODS
General information
This is a prospective observational study, including ten sepsis patients (sepsis group)admitted to the ICU of the General Hospital, Ningxia Medical University, China from February 2017 to June 2017 , ten patients without sepsis (non-sepsis group) admitted to the same ICU during the same time period, and ten local healthy individuals (control group). The inclusion criteria for sepsis patients were as follows: 18 -75 years old;sepsis patients meeting the latest definition of sepsis in “Sepsis-3 .0 ” issued by the Society of Critical Care Medicine (SCCM) in 2016 [7 ]; and estimated time of more than 2 d spent in the ICU after enrollment. The inclusion criteria for the non-sepsis patients were as follows: 18 -75 years old; and patients admitted to the ICU at the same period owing to diseases other than sepsis, such as multiple injuries and high-risk operations.Of note, we tried to match the age, underlying disease, and surgical site with those in patients in the sepsis group. Because there was no definite infection, the non-sepsis group was not given antibiotics before the fecal samples were collected. The exclusion criteria were as follows: Not meeting the inclusion criteria; perianal infection; patients subjected to enterostomy; and patients with chronic gastrointestinal diseases.Additionally, the requirements for the healthy control group were as follows: Matched age with the aforementioned two groups of patients; good health; no history of chronic or metabolic diseases (e.g., hypertension, coronary heart disease, diabetes, hepatitis,and hyperthyroidism); no history of digestive tract diseases or digestive tract surgery;and not using antibiotics, probiotics, enteral nutrients, or other drugs within 3 mo before enrollment. Informed consent was obtained from all patients or their immediate family members, and this study was approved by the hospital ethics committee (ethics approval number: 2016 -258 ).
Research methods
The general clinical data of patients in the sepsis and non-sepsis groups were recorded, including age, main diagnosis, operation type (abdominal cavity organ operation and non-abdominal cavity organ operation), clinical infection site,pathogenic bacterial agent (based on qualitative culture), and use of antibiotics (Tables 1 -3 ). The acute physiology and chronic health (APACHE II) score and sequential organ failure assessment (SOFA) score of patients on the day of ICU admission were recorded (Table 1). The primary infection site, surgical site, and use of antibiotics in the sepsis group were also recorded (Table 2), as were the positive results of blood, urine,sputum, and other body fluids collected from patients with sepsis (Table 3).Additionally, venous blood samples were collected from patients with sepsis on days 1 , 3 , and 7 after admission to the ICU, and procalcitonin (PCT) was detected by immunochromatography [PCT-Q detection card produced by BRAHMS GmbH in Germany, provided by Thermo Fisher Scientific (China) Co., Ltd.], while the levels of serum D-lactic acid (D-Lac), bacterial endotoxin (endotoxin), and diamine oxidase(DAO) were determined using the JY-DLT intestinal barrier function biochemical index analysis system (Beijing Zhongsheng Jinyu Diagnosis Technology Co., Ltd.,China.) within 0 .5 h after blood collection.
Collection of stool samples and sequencing
Stool samples were collected from sepsis patients on days 1 , 3 , and 7 after admission to the ICU, from non-sepsis patients on day 1 after admission to the ICU, and from healthy controls. Samples from patients were taken from a deep part of the fresh stool of patients by using a sampling spoon, and the stool samples were quickly placed in a special stool sample box. The stool samples of healthy control subjects were collectedin a special stool sample box after normal defecation. Then, the small stool boxes filled with samples were sealed, labeled, placed in a liquid nitrogen tank, and transferred to a -80 °C freezer for storage.
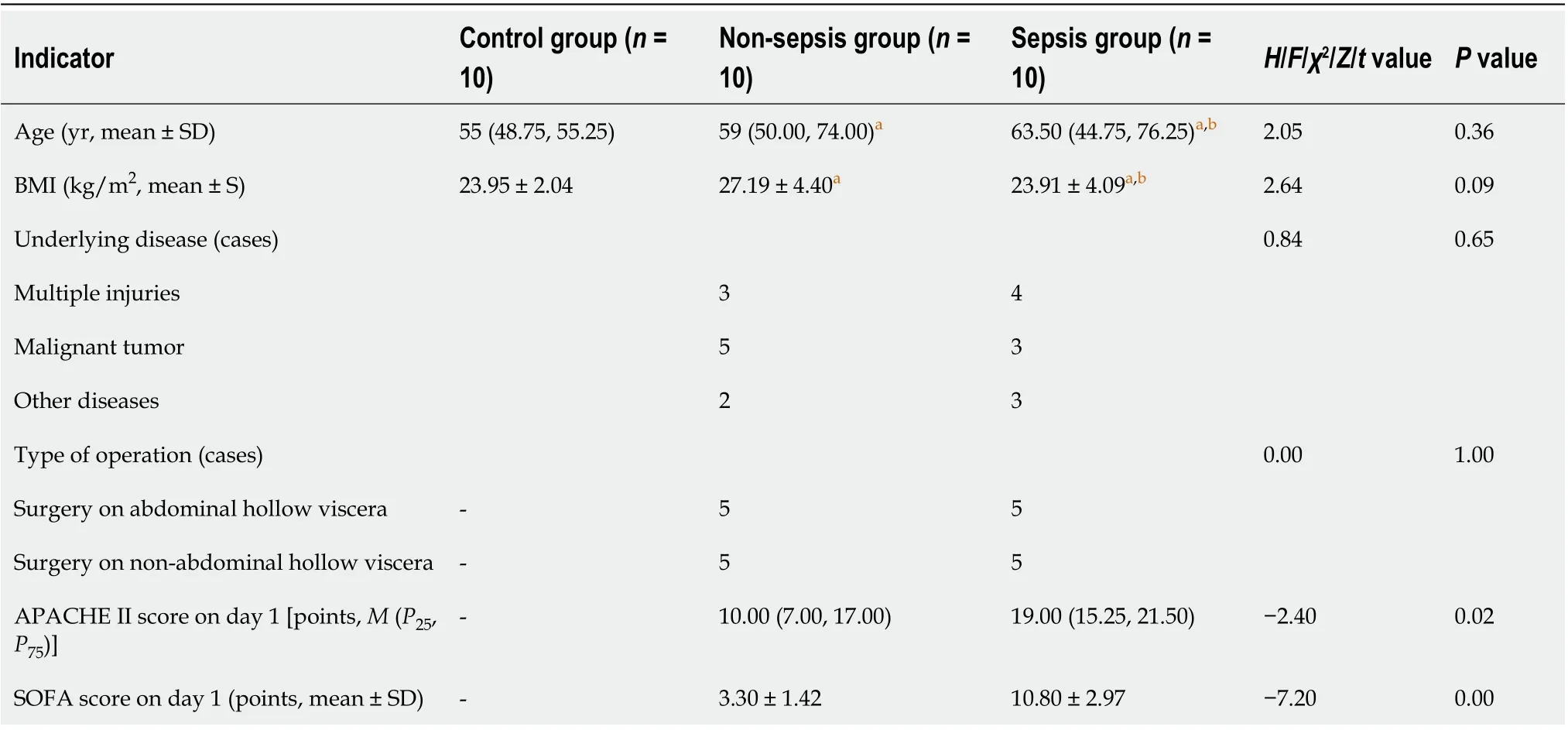
Table 1 Comparison of the general information among the three groups of individuals
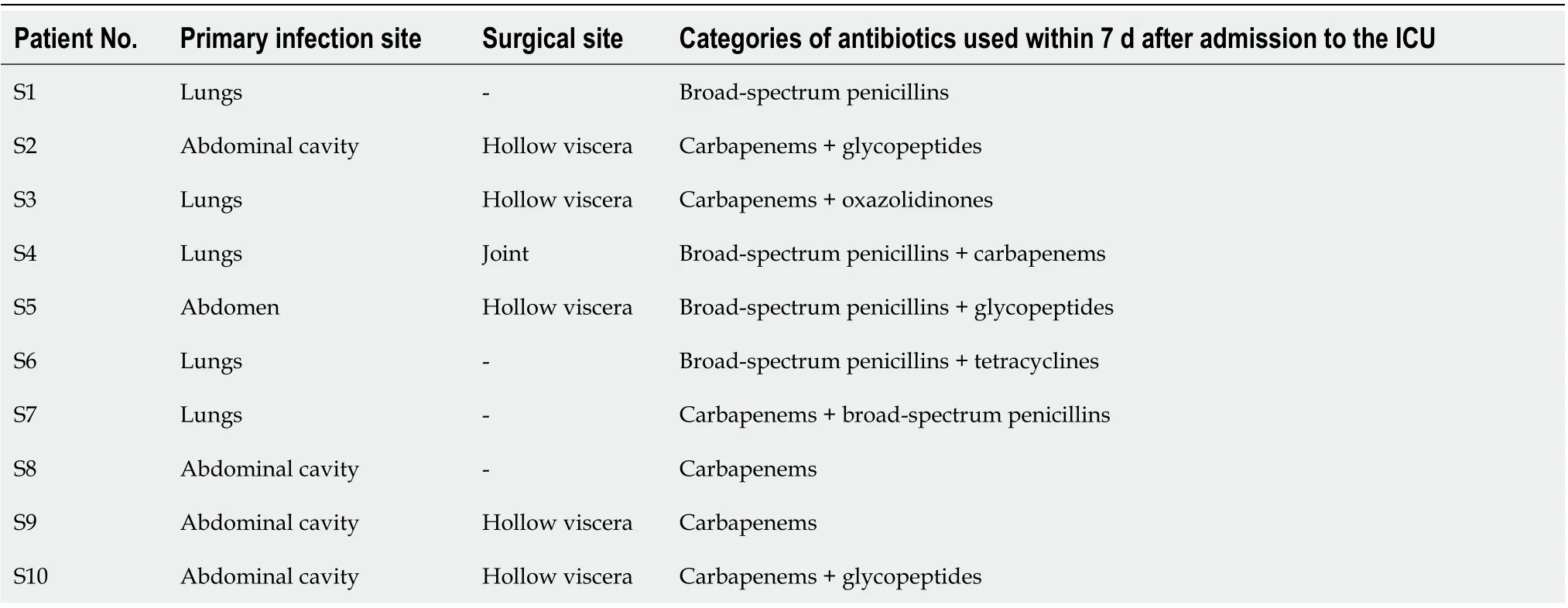
Table 2 General clinical information of the patients in the sepsis group
Each stool sample was added to 790 μL of lysis buffer [4 M guanidine thiocyanate,250 μL; 10 % N-lauroyl sarcosine, 40 μL; 5 % N-lauroyl sarcosine-0 .1 M phosphate buffer (pH 8 .0 ), 500 μL] together with 1 g glass beads (0 .1 mm, Biospec Products, Inc.,United States). After sufficient vortex-based homogenization, bead beating was performed for 10 min at full speed. The subsequent extraction was carried out according to the instructions of the extraction kit manufacturer (E.Z.N.A.®Stool DNA Kit, Omega Bio-tek, Inc., GA). The V3 –V4 region of the 16S rRNAgene was amplified with the primers 341 F/805 R (341 F: 5 ′-CCTACGGGNGGCWGCAG-3 ′; 805 R: 5 ′-GACTACHVGGGTATCTAATCC-3 ′), and the PCR products were sequenced on an Illumina MiSeq 2 *300 bp platform. The raw sequencing data and accompanying information are available in the Sequence Read Archive database under the accessionnumber PRJNA691455 .
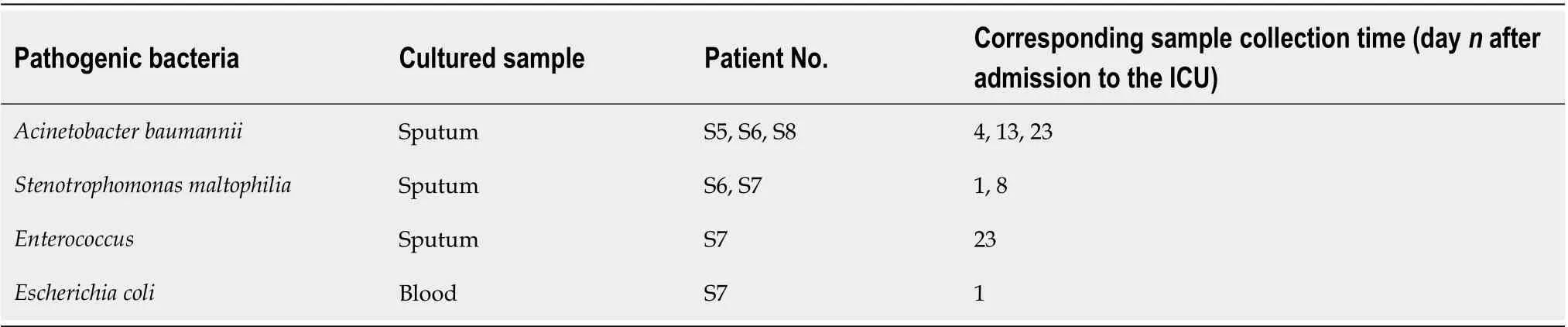
Table 3 Pathogenic bacteria isolated in the sepsis group
Bioinformatics and statistical analysis
The clinical data were processed and analyzed using SPSS 19 .0 software. All data were first tested for normality and homogeneity of variance. Data conforming to a normal distribution and homogeneity of variance are expressed as the mean ± SD, and thettest or analysis of variance were used for statistical analysis. Data not conforming to a normal distribution are expressed as medians (P25 , P75 ), and the rank-sum test was used for statistical analysis. Theχ2test was used to compare enumeration data, and P < 0 .05 was considered statistically significant.
USEARCH 8 .0 was used to process the raw sequencing data, and operational taxonomic units (OTUs) were classified according to 97 % sequence similarity. The OTU representative sequences were compared against the SILVA database (SSU123 ;http://www.arb-silva.de) with a 70 % confidence threshold, and the taxonomic status of each16S rDNAsequence was obtained. The α-diversity of each sample was evaluated using the Shannon–Wiener and Simpson’s diversity indexes, and the significance of differences was tested using either the nonparametric Mann–WhitneyUtest or the Kruskal–Wallis rank-sum test. Principal coordinates analysis (PCoA) and the calculation of the linear discriminant analysis effect size (LEfSe) were performed using the R software package (http://www.R-project.org/); heatmaps were also obtained using the R software. Spearman correlation analysis was also carried out to assess the relationships between relevant parameters.
RESULTS
Characteristics of the participants
A total of ten sepsis patients, ten non-sepsis patients, and ten healthy individuals were included in this study. Table 1 shows that there were no significant differences with respect to age or body mass index among the groups, as revealed by variance analysis(P> 0 .05 ). The underlying diseases of the patients in the sepsis and non-sepsis groups were divided into three categories: Multiple injuries, malignant tumors, and other diseases (including benign tumors and intestinal obstruction). Of note, there were no significant differences in the underlying diseases and operation types between the sepsis and non-sepsis groups as revealed by theχ2test (P > 0 .05 ), indicating that the groups were matched (and accurate comparisons were possible). The APACHE II and SOFA scores in the sepsis group were significantly higher than those in the non-sepsis group on day 1 of ICU admission (P < 0 .05 ), suggesting that the patients in the sepsis group had worse pathology (with acute organ dysfunction) compared with those in the non-sepsis group.
Comparison of intestinal flora structure in healthy individuals and non-sepsis and sepsis patients on day 1 after ICU admittance
A total of 49 fecal samples were included for analysis. Of note, one stool sample in the sepsis group (patient S3 , day 3 ) was missing owing to a problem in specimen collection. After quality control procedures, 629665 high-quality sequences were obtained, and the qualified reads were clustered into 440 species-level OTUs using 97 % as the similarity cutoff. In this study, the number of qualified reads of inpatients in the ICU was generally much lower than that of healthy controls. To understand whether these differences were due to low bacterial numbers, we randomly selected five specimens from each group to quantify the total bacteria in the feces. Regardless of whether bacteria were counted as bacterial copy number per gram fecal samples(mean bacterial copy numbers in sepsis, non-sepsis, and healthy control groups, 5 .07 E+ 11 , 1 .36 E + 12 , and 9 .27 E + 12 , respectively) or per nanogram bacterial DNA (9 .02 E +06 , 3 .54 E + 07 , and 4 .90 E + 07 , respectively), there was a significant overall decreasing trend in both the sepsis and non-sepsis groups relative to that in healthy controls.Therefore, altogether, these results suggest that the low number of qualified reads in some samples in the sepsis and non-sepsis group is secondary to the significantly reduced number of intestinal bacteria, probably because of the use of large amounts of antibiotics in the ICU.
α-diversity:The Mann–Whitney test showed that the α-diversity in the non-sepsis and sepsis groups on day 1 after ICU admittance was significantly lower than that in the control group (P< 0 .05 ). Moreover, the diversity of the intestinal flora in sepsis patients was lower than that in non-sepsis patients; however, this difference was not significant (Figure 1 and Table 4 ).
Overall structural comparison of the intestinal flora:The PCoA plot based on the Bray–Curtis dissimilarity between samples revealed that the flora composition in sepsis patients was similar to that in non-sepsis patients on day 1 after ICU admittance(Figure 2). In line with these results, Adonis analysis showed no significant difference in the composition of the intestinal flora between sepsis and non-sepsis patients on day 1 after ICU admittance (P > 0 .05 ). However, the difference in the flora composition between these groups and normal controls was extremely significant (AdonisP<0 .05 ).
Comparison of intestinal flora compositions at the phylum level:The flora composition results revealed that Firmicutes and Bacteroidetes were the dominant phyla in healthy individuals, accounting for more than 70 % of the total bacteria.Meanwhile, in sepsis and non-sepsis patients, Firmicutes was the dominant phylum in the intestinal flora (Figure 3 A); the proportion of Bacteroidetes decreased significantly compared to that in healthy individuals (P< 0 .05 ), and the relative abundance of Proteobacteria increased compared to that in normal controls, although the difference was not significant. Of note, the proportion of Fusobacteria in the intestinal tract of non-sepsis patients was significantly higher than that in both healthy controls and sepsis patients (P< 0 .05 ; Figure 3 B).
Comparison of intestinal flora compositions at the genus and OTU levels:The results of the Kruskal–Wallis test showed that the proportions ofPrevotella,
Subdoligranulum,Lachnospira,Phascolarctobacterium,Alloprevotella,Megamonas, andParasutterellain the intestinal tract of patients in the non-sepsis and sepsis groups decreased significantly compared to those in the healthy control group (P< 0 .05 ).Furthermore, the abundance ofEnterococcuswas much higher in the intestinal tract of sepsis patients than in non-sepsis patients and healthy controls. In contrast, the abundance ofFusobacterium,Anaerococcus, andPeptostreptococcuswas significantly higher in the intestinal tract of non-sepsis patients than in healthy controls and sepsis patients (Figure 4 A).
Additionally, as per the heatmap analysis (random forest-based), 64 key OTUs were different among the three groups (Figure 4 B). Among these, the relative abundance of OTUs assigned to the generaSubdoligranulum,Alistipes,Megamonas,Blautia, Bacteroides,Dialister,Anaerostipes,Lachnospira,Roseburia,Pseudobutyrivibrio, Parasutterella, Prevotella,Coprococcus,Parabacteroides,Paraprevotella, andBifidobacteriumwas lower in the nonsepsis and sepsis groups than in the normal control group, whereas the abundance of OTUs assigned to the generaPeptostreptococcus,Enterococcus,Thermoanaerobacterium,andAnoxybacilluswas decreased in the intestinal microbiota of the normal control group. In addition, the relative abundance of OTUs assigned to the generaGranulicatellaandStreptococcuswas lower in the sepsis group than in the non-sepsis group.
Dynamic changes in intestinal flora structure in sepsis patients undergoing treatment
α-diversity:The Mann–Whitney test showed that the α-diversity in the sepsis group decreased significantly within 1 wk (from days 1 to 7 after the initiation of ICU treatment) compared to that in the control group; importantly, these differences were significant (P< 0 .05 ). Of note, on days 1 , 3 , and 7 of treatment, the α-diversity of the intestinal flora decreased gradually, but pairwise comparisons did not showsignificant differences. This suggests that antibiotics killed many intestinal bacteria,and that the diversity of the intestinal flora not only did not recover but also showed a downward trend when antibiotics were continuously used (Figure 5 and Table 5 ).
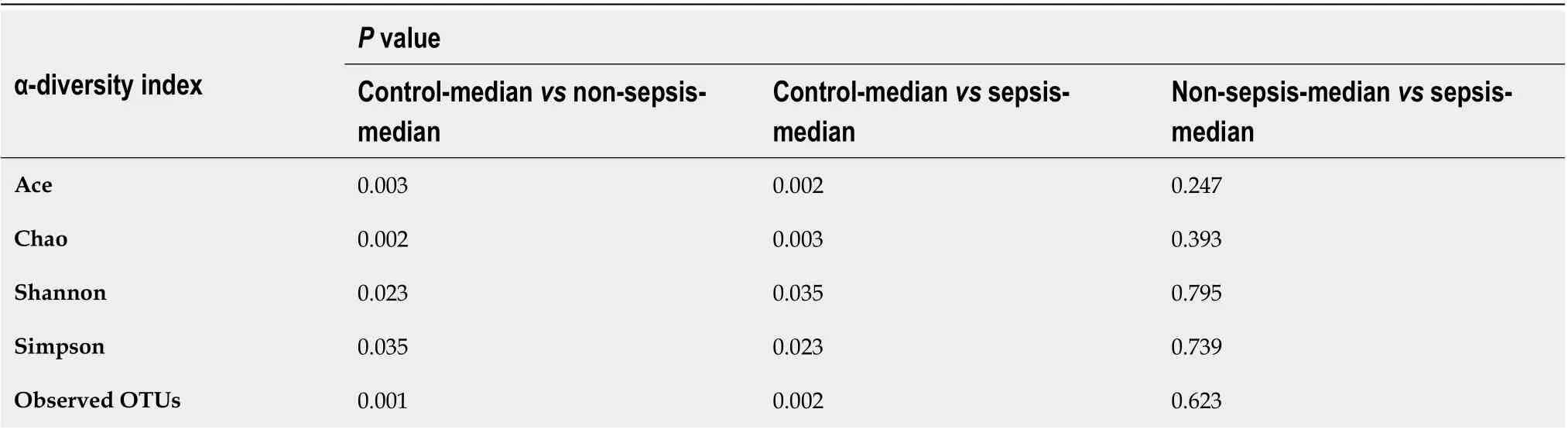
Table 4 Statistical comparison of the α-diversity indexes of the intestinal flora in the three groups of individuals
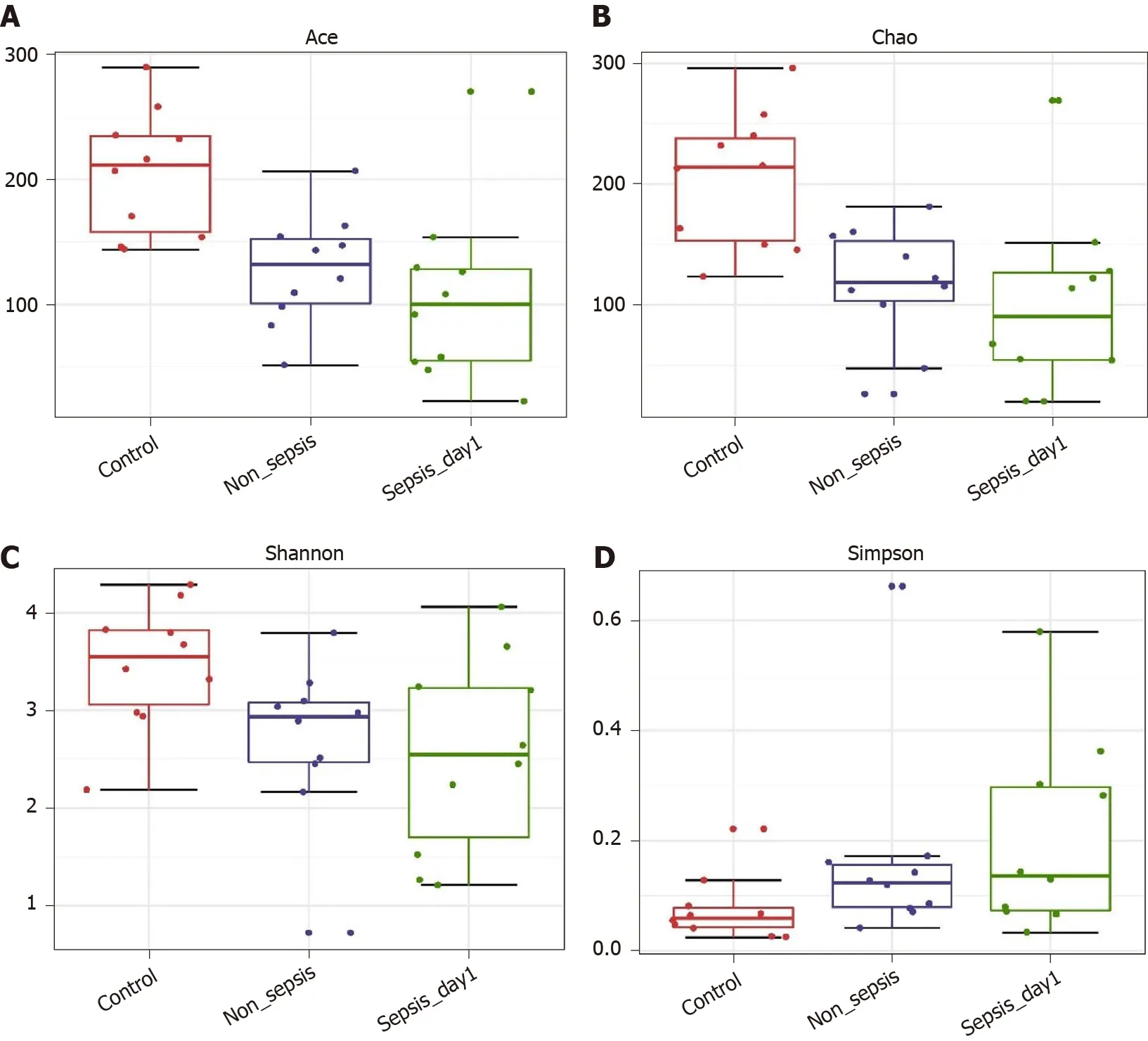
Figure 1 α-diversity indexes of fecal microbiota in normal individuals, non-sepsis patients, and sepsis patients. A: ACE estimator; B: Chao 1 estimator; C: Shannon index; D: Simpson index.
Overall intestinal flora composition changes in sepsis patients within 1wk after treatment:The PCoA plot based on Bray–Curtis dissimilarities between samples showed that the intestinal flora composition in sepsis patients within 1 wk of treatment (from days 1 to 7 after ICU admittance) was different from that in normal controls (Figure 6); Adonis analysis showed that the difference between sepsis patientsand normal controls was extremely significant (P< 0 .05 ). However, the structure of the intestinal flora of sepsis patients did not differ significantly with respect to the different days after ICU admittance in the sepsis group.
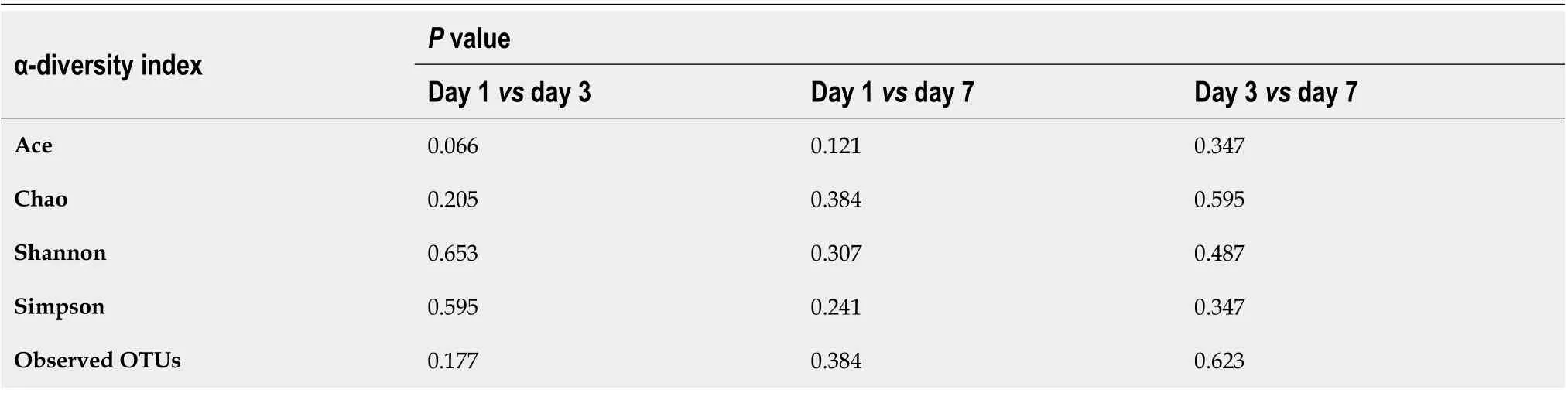
Table 5 Variation in the α-diversity indexes of the intestinal flora in patients with sepsis within 1 wk of treatment (days 1 , 3 , and 7 after intensive care unit admittance)
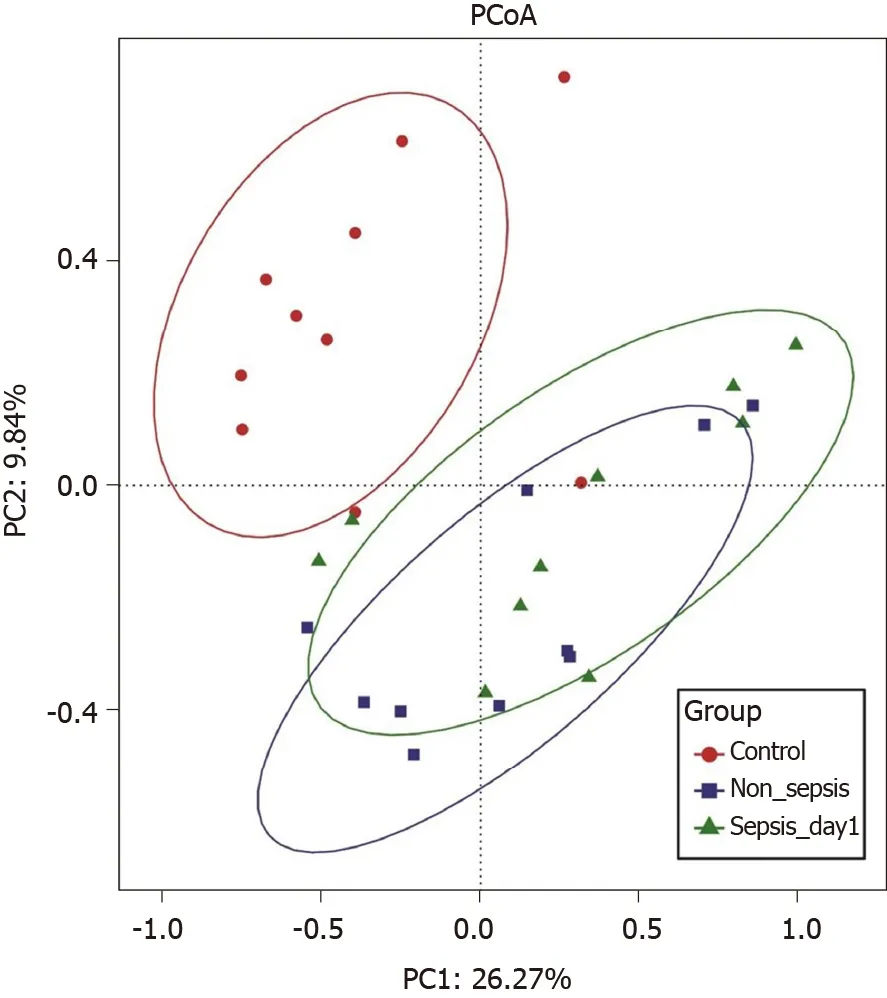
Figure 2 Principal coordinate analysis of intestinal flora among normal individuals, sepsis patients, and non-sepsis patients. Principal coordinate analysis was plotted based on the Bray–Curtis dissimilarity between samples. The ellipses highlight the clustering of the fecal microbiomes according to groups (red: Healthy control group; blue: Non-sepsis group; green: Day 1 of sepsis).
Identification of differential bacterial taxa between sepsis patients 1wk after treatment and normal individuals:To screen for the bacterial genera associated with significant differences in the intestinal flora of sepsis patients within 1 wk of admission to the ICU, we reduced the dimensionsviathe calculation of LEfSe to obtain the linear discriminant analysis scores. Compared to normal controls, in sepsis patients, some harmful bacteria such asCoprococcusdisappeared from the intestinal flora within 1 wk of ICU treatment, indicating the efficacy of drugs against pathogenic bacteria(Figure 7). At the same time, the abundance of most of the beneficial bacteria, such asPrevotellaandBifidobacterium, also decreased, indicating that the effect of drug treatment, especially antibiotics, was not limited to harmful bacteria. However, the abundance of some bacteria, such asEnterococcusandHemophilus, still increased despite the action of antibiotics, indicating that these bacteria were resistant to the antibiotics used.
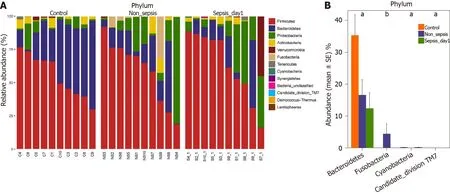
Figure 3 Composition and comparison of the fecal microbiota among normal individuals, sepsis patients, and non-sepsis patients at the phylum level. A: Composition of the fecal microbiota at the phylum level among three groups; B: Comparison of the fecal microbiota at the phylum level among three groups. The average abundance of each phylum is depicted as the mean ± SE. P values were calculated using the Kruskal–Wallis rank-sum test: aP < 0 .05 , bP< 0 .01 .
Correlation analysis between clinical indicators and abundance of intestinal bacterial genera in patients with sepsis
There were ten patients in the sepsis group (S1 –S10 ). Venous blood samples were collected on days 1 , 3 , and 7 after admission to the ICU to determine the levels of permeability-related D-Lac, endotoxin, and DAO and infection-related PCT. In the blood of sepsis patients, the levels of D-Lac, endotoxin, DAO, and PCT were 20 .96 ±11 .90 mg/L, 10 .65 ± 7 .92 U/L, 19 .58 ± 17 .61 U/L, and 7 .23 ± 13 .92 ng/mL, respectively.
Based on the Spearman correlation analysis, significant correlations were established between clinical indicators and abundance of intestinal bacterial genera in sepsis patients (Figure 8). The number of days of admission in the ICU negatively correlated with the abundance ofStreptococcusandRuminococcus(P< 0 .05 ). The abundance ofRoseburiaandParasutterellawas positively correlated (P< 0 .05 ), while that ofPrevotella,Lactobacillus, andFinegoldiawas negatively correlated with the levels of D-Lac (P< 0 .05 ). Additionally, the abundance ofRuminococcus,Roseburia,Sutterella,
Peptostreptococcus,Escherichia-Shigella,Dorea,Bilophila,Coprobacter,Clostridium_sensu_stricto_1,Parabacteroides,Bifidobacterium,Alistipes, andParasutterella, as well as some genera belonging to Lachnospiraceae, Christensenellaceae, Ruminococcaceae, and Erysipelotrichaceae, was positively correlated with the levels of PCT (P< 0 .05 ).Conversely, the abundance ofStenotrophomonasandEnterococcuswas negatively correlated with the levels of PCT (P< 0 .05 ). The abundance ofPeptostreptococcus,Roseburia,Collinsella,Dorea, and VadinBB60 group norank was also positively correlated with the endotoxin levels (P< 0 .05 ), while that ofDialister,Pepto streptococcus, with the endotoxin levPeptostreptococcus,Dorea,Hemophilus,Bifidobacterium,Collinsella,andBlautiawas positively correlated with the levels of DAO(P< 0 .05 ).
DISCUSSION
Characteristics of intestinal flora structure in sepsis patients
Sepsis is associated with high morbidity and mortality worldwide, and its timely identification and treatment are difficult. Therefore, improving the diagnosis and treatment of sepsis is essential from a global health perspective[8 ]. In February 2016 ,the SCCM and the European Society of Intensive Care Medicine jointly released the definition of sepsis in “Sepsis 3 .0 ” as follows: “Life-threatening organ dysfunction due to a dysregulated host response to infection”[7]. This definition emphasizes the close relationship between sepsis and organ dysfunction; of note, the intestinal tract is recognized as the “engine” of organ dysfunction. The intestinal flora, as an important component of the intestinal tract, plays major physiological roles in the context of biosynthesis and metabolism; however, critical diseases lead to many changes in the diversity of the intestinal flora, causing excessive growth of pathogenic bacteria[9 ,10 ].
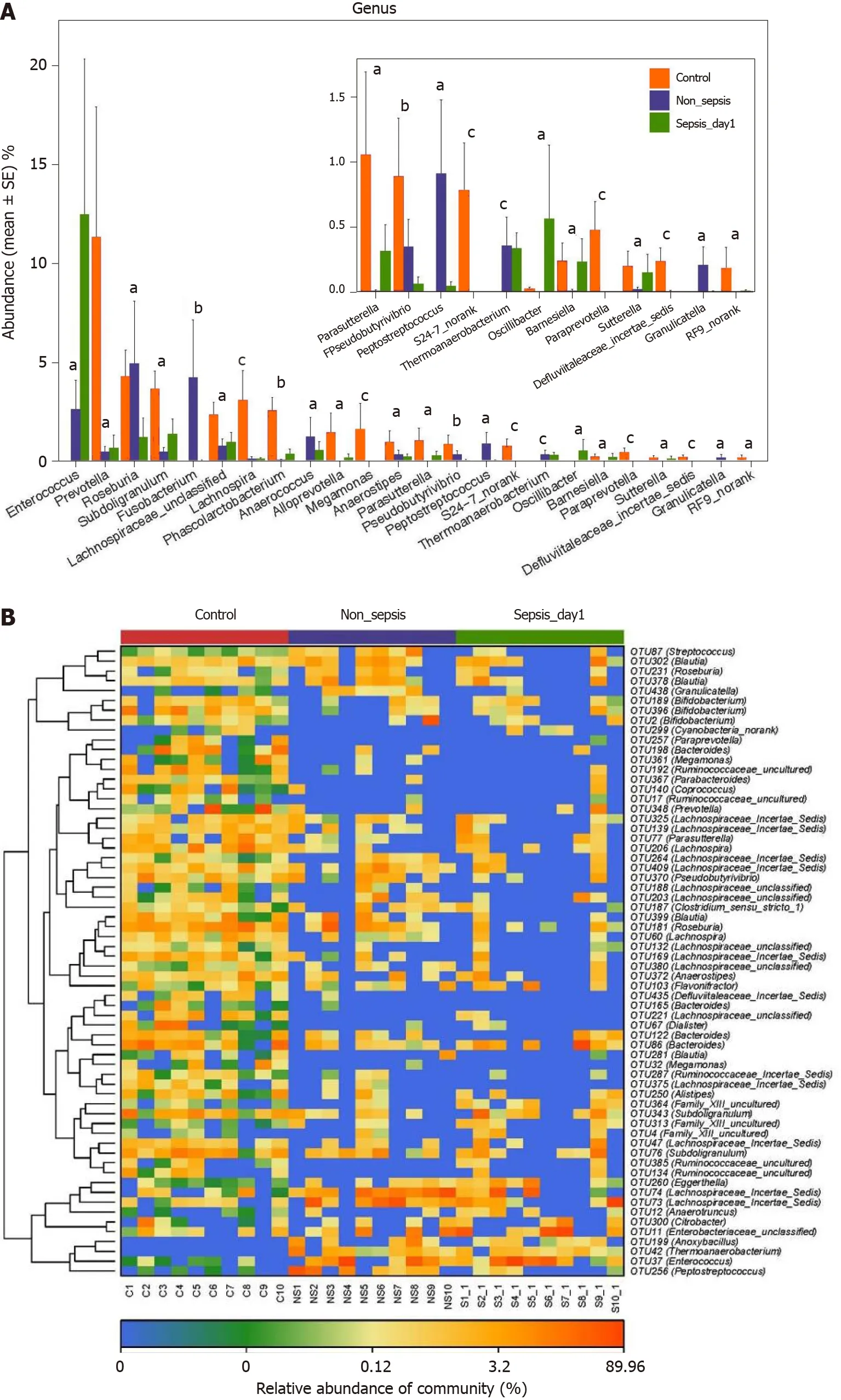
Figure 4 Key phylotypes showing significant differences among normal individuals, sepsis patients, and non-sepsis patients. A:Comparison of the fecal microbiota at the genus level among the three groups. The average abundance of each genus is depicted as the mean ± SE. P values were calculated using the Kruskal–Wallis rank-sum test: aP < 0 .05 ; bP < 0 .01 , cP < 0 .001 ; B: Heatmap representing the relative abundance of differential operational taxonomic units (OTUs) among the three groups. The key OTUs whose relative abundance was above 1%, as per random forest-based models are displayed in the“figure”.
By comparing the intestinal flora of sepsis patients and healthy controls, we confirmed that there are marked differences in the abundance, distribution, and structure of the intestinal flora in the context of sepsis. In sepsis patients, normal bacteria decreased or disappeared, while the abundance of abnormal bacteria increased sharply. At the phylum level, the abundance of Firmicutes increased significantly, while that of Bacteroidetes decreased significantly in the intestinal flora of sepsis patientsvsnon-septic critically ill patients; of note, the number of bacteria in the phylum Fusobacterium also decreased significantly. At the genus level, in addition to the decrease in the abundance of beneficial symbiotic bacteria such asPrevotellaandLachnospira, as well as of other genera, includingFusobacteriumandPeptostreptococcus,we found that the abundance ofEnterococcusincreased significantly in the intestinal flora of sepsis patientsvsnon-septic critically ill patients. Therefore, our results suggest that dysbiosis in sepsis shows three major features. First, as the abundance and diversity of the intestinal flora decrease, the structure of the intestinal flora changes, and the differences among individuals are greater[11 ]. Second, the abundance of dominant obligate anaerobes decreases and that of facultative anaerobes increases[11 ,12 ]. Third, the abundance of beneficial symbiotic bacteria decreases, while that of pathogenic bacteria increases, possibly becoming predominant[11 ,13 ].
The occurrence of intestinal microecological disorders in sepsis patients is not surprising. First, the physiological state of sepsis patients is completely different from that at homeostasis; specifically, intestinal hypoperfusion and reperfusion injury lead to changes in the intestinal environment and blood supply to the intestinal mucosa,resulting in intestinal mucosal inflammation and, consequently, a series of changes in the intestinal environment, such as increased nitrate concentrations[11 ] and altered mucosal oxygen gradient[12 ]. These changes are favorable to the growth of Proteobacteria, leading to the expansion of many clinically familiar pathogenic Gramnegative bacilli, such asPseudomonas aeruginosaandEscherichia coli, as well asStaphylococcus aureusandEnterococcus[14 ,15 ]. Second, ICU patients are exposed to various endogenous regulators (such as increased catecholamine production and changes in glucose metabolism) and clinical interventions (such as proton pump inhibitors,opioids, nutritional support, and antibiotics), which affect the living environment of the intestinal flora to varying degrees and, thus, affect the flora structure[16 ]. Finally,the intestinal mucosa will be damaged and thinned in critically ill patients[17 ,18 ]; such mucosal damage leads to the loss of the normal habitat of symbiotic microorganisms,subsequently leading to intestinal microecological disorders.
Dynamic changes in the structure of the intestinal flora of sepsis patients within 1 wk after treatment initiation
Our study suggests that the abundance, distribution, and diversity of the intestinal flora in sepsis patients do not change significantly within 1 wk of ICU admittance.However, in relation to the gut microbiome of healthy subjects, the numbers of most beneficial bacteria, such asPrevotellaandBifidobacterium, were below the detection limit in sepsis patients, whereas the abundance of infection-related bacteria, such asEnterococcusandHemophilus, increased despite the action of antibiotics (with the exception ofCoprococcus, which decreased in abundance to below the detection limit).These results indicate that patients with intestinal flora disorders associated with sepsis could not recover in a short time, and while the drugs were effective against the bacteria causing infection, they also affected bacteria in the intestines. In fact, the effects of drug treatment, especially antibiotics, on bacteria were not limited to harmful bacteria; the proportion of beneficial intestinal bacteria also decreased. Other studies have also shown that the long-term use of antibiotics changes the normally healthy intestinal flora and leads to the emergence of drug resistance; worrisome enough, the long-term use of antibiotics has the potential to generate organism reservoirs with a multi-drug resistance gene pool[19 ]. Ma et al[20 ] used rat models with third-degree burns on 30 % of the total surface area of the back and quantified/identified the intestinal bacteria after treatment. The results showed that the number of cocci in the gastrointestinal contents of rats increased significantly, and the coccus/bacillus ratio was seriously inverted after treatment with Rocephin. It was considered that broad-spectrum antibiotics destroyed the intestinal microecological balance; of note, the conditional pathogenic intestinal flora showed potential to affect health, disease, and drug action. Our study also revealed that the abundance ofEnterococcusincreased significantly in sepsis patients, which might be related to the use of broad-spectrum antibiotics.
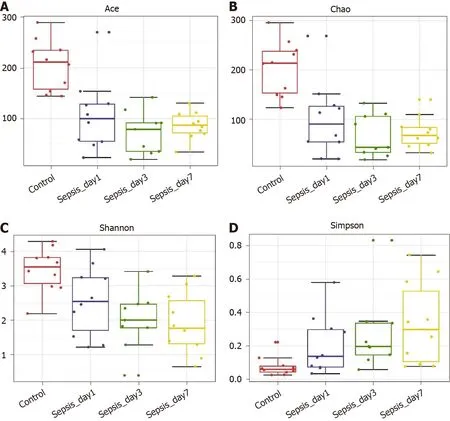
Figure 5 α-diversity indexes of the fecal microbiota in sepsis patients on days 1 , 3 , and 7 after admission to the intensive care unit vs healthy individuals. A: ACE estimator; B: Chao 1 estimator; C: Shannon index; D: Simpson index.
Correlation between the abundance of intestinal bacterial genera and clinical indicators in patients with sepsis
Our study showed a clear correlation between the abundance of intestinal flora components and clinical indicators in sepsis. The clinical indicators used in this study were D-Lac, endotoxin, DAO, and PCT. Under normal circumstances, D-Lac is producedviathe methylglyoxal metabolism, and its content in the blood is very small.However, when glycolysis results in increases in a large number of gastrointestinal bacteria and the intestinal barrier function is impaired, the content of D-Lac increases sharply. Endotoxins are cell wall components of Gram-negative bacteria that are released only when bacteria are lysed and die. DAO is a highly active intracellular enzyme in the upper villi of the intestinal mucosa of mammals, including humans. Its activity is closely associated with nucleic acid and protein synthesis in mucosal cells.Therefore, the above three indicators can reflect the integrity and damage degree of the intestinal mechanical barrier. Additionally, PCT reflects the active level of the systemic inflammatory response, and the factors affecting its levels include the size and type of the infected organ, the type of pathogenic bacteria, the degree of inflammation, and the state of immune responses. Our study showed that serum PCT, endotoxin, DAO,and D-Lac levels in patients with sepsis were correlated with the abundance of various intestinal bacterial genera. Of note, some of these genera were correlated with multiple clinical indicators at the same time. For example, the abundance ofPeptostreptococcusandDoreawas positively correlated with the serum levels of PCT, endotoxin, and DAO; moreover, the abundance ofRoseburiawas positively correlated with the serum levels of PCT, endotoxin, and D-Lac.
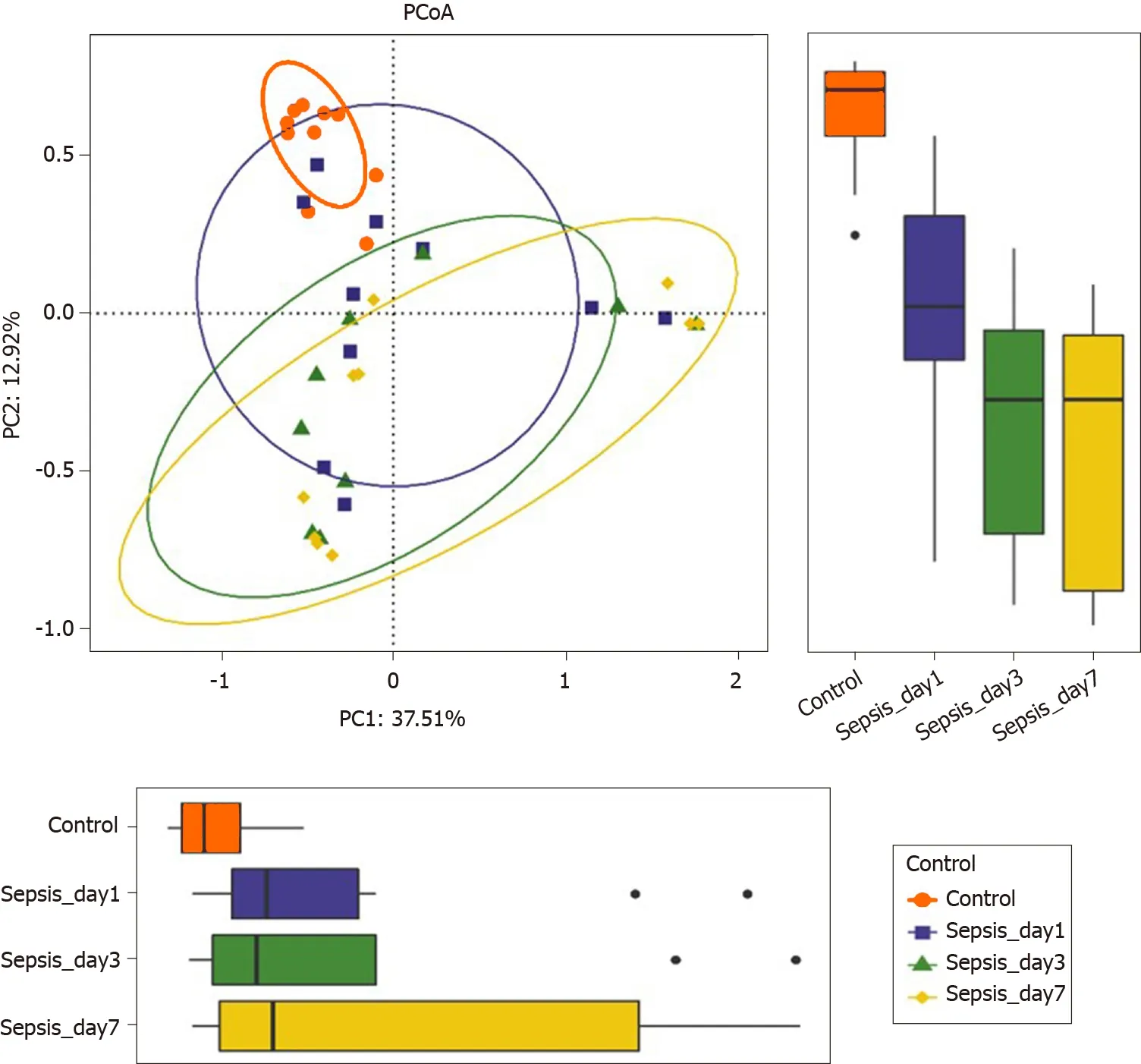
Figure 6 Principal coordinate analysis of the intestinal flora in sepsis patients on days 1 , 3 , and 7 after admission to the intensive care unit versus healthy individuals. Principal coordinate analysis was plotted based on the Bray–Curtis dissimilarity between samples. The ellipses highlight the clustering of the fecal microbiomes according to the groups (red: Healthy control group; blue: Sepsis patients on day 1 ; green: Sepsis patients on day 3 ; yellow:Sepsis patients on day 7).
Among these genera, the abundance ofRuminococcushad the highest positive correlation with PCT.Ruminococcusis an important constituent of the normal intestinal microbiota. A study published in 2017 [21 ] using samples from young Han Chinese individuals revealed changes in the intestinal flora of the Chinese population and showed that the generaRuminococcusandFusobacterium, among others, were relatively highly abundant in obese Chinese individuals, whereas the abundance ofBacteroideswas greatly reduced. Of note, twoRuminococcusspecies,Ruminococcus torquesandRuminococcus gnavus, should be mentioned because they are related to inflammatory bowel disease[22 ] and metabolic disorder[23 ]. Importantly, from the significant decrease in the number and abundance of intestinal bacteria, including the decrease in abundance of the phylum Bacteroidetes, the increase in abundance of the phylum Verrucomicrobia, and the increase in abundance of the genusRuminococcusin sepsis patients, we can establish a parallel with the results of the study of obese Chinese individuals. In clinical practice, it is known that obese individuals have more significant characteristics of insulin resistance and dyslipidemia, with a more significant inflammatory phenotype, which are risk factors for cardiovascular diseases,diabetes, osteoporosis, and some cancers among other diseases[24 ]. Interestingly,patients with sepsis also have clinical manifestations such as insulin resistance, dyslipidemia, and inflammatory responses, which have adverse effects on the severity of illness and prognosis[25 ]. Therefore, we boldly speculate that the intestinal phenotype of obese patients makes them more susceptible to the complications of sepsis, which might be of great significance for the treatment and prevention of sepsis. Of course, the sample size in this study was small, and thus, it is necessary to conduct larger,multicenter studies to support our findings; moreover, the proposed mechanisms require further validation in animal studies for clarification.
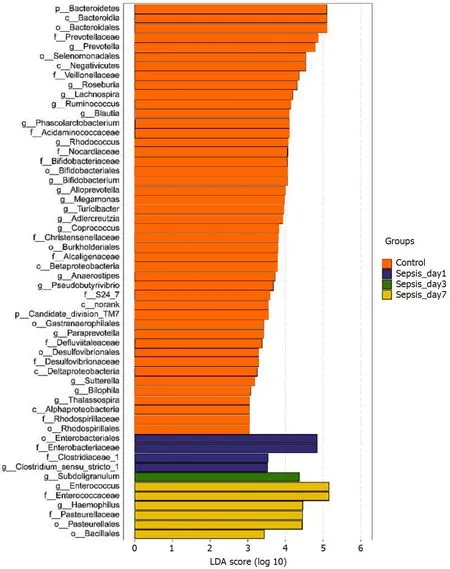
Figure 7 Linear discriminant analysis scores indicating significant differences in the fecal microbiota between the sepsis patients within 1 wk after admission to the intensive care unit and the normal control group. Red: Enriched taxa in the healthy control group; blue: Enriched taxa in sepsis patients on day 1 ; green: Enriched taxa in sepsis patients on day 3 ; yellow: Enriched taxa in sepsis patients on day 7 .
There is a correlation between intestinal flora disorders and intestinal barrier dysfunction in sepsis patients. Our research showed that the abundance ofRoseburiain the intestinal tract of sepsis patients was definitely related to the levels of serum markers of intestinal barrier function.Roseburiais one of the main bacterial genera producing butyrate in the human intestinal flora[26 ,27 ]. Butyrate is the main energy source of colonic epithelial cells. Research suggests that the abundance of bacteria producing butyrate in the intestinal tract of critically ill patients decreases or disappears, which leads to a decrease in butyrate production and the apoptosis of intestinal epithelial cells due to “starvation”[26 ]. Importantly, one study showed that the genusRoseburiacan improve the intestinal ecosystem, prevent intestinal leakage,and reduce the incidence of diabetes[27 ]. Conversely, other studies have shown that the abundance ofRoseburiais significantly reduced in patients with Crohn’s disease[28 ] and inflammatory bowel disease[29 ]. Our study showed that the abundance of intestinalRoseburiawas positively correlated with serum PCT,endotoxin, and D-Lac levels in sepsis patients, indicating that the intestinal barrier function is more severely damaged and the systemic inflammatory response is more serious with a higher content ofRoseburia, which seems to contradict the results of existing research. We speculate that systemic inflammatory responses in sepsis patients cause intestinal damage and that the body has a self-regulatory effect on this damage. In addition, a recently published study[30 ] showed that immune cells and autoantibodies of patients with antiphospholipid syndrome (an autoimmune disease)could cross-react with the mimic epitopes ofRoseburia; in line with this,Roseburiacould also cause autoimmune diseases in susceptible mice. Once again, these results suggest that the positive and negative effects of intestinal bacteria are not invariable.
Bacteria can perceive the changes in the host internal environment and then change their own toxic factors to become pathogenic. Under various stimuli such as sepsis,trauma, and burns, a healthy bacterial strain might be transformed into a pathogenic bacterium within a few hours[31 ]. For example,Pseudomonas aeruginosawas injected into the ceca of mice in a sham operation group for culture, and then, the bacteria were collected and implanted into the undamaged abdominal cavity of mice; it was found that no death occurred. However, the same strain was injected into the ceca of mice after 30 % hepatectomy for culture, and then were collected and implanted into the undamaged abdominal cavities of mice, which led to their deaths[32 ]. Although the mechanisms underlying such transformations remain inconclusive, these results suggest that the host environment can not only change the diversity of bacterial species but also change their virulence. Therefore, the beneficial or harmful effects of a specific bacterium are not absolute; they can be influenced by many factors.
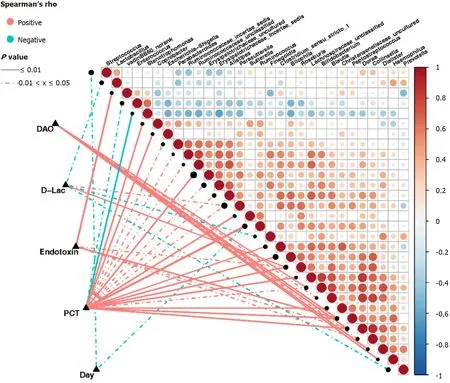
Figure 8 Correlation between clinical indicators and abundance of fecal microbiota in sepsis patients. Heatmap showing partial Spearman’s correlation coefficients among 30 genera and clinical indexes. Connecting lines represent the correlation coefficient values above 0 .4 (red, positive correlation) or below -0 .4 (blue, negative correlation). Solid lines represent P ≤ 0 .01 . Dotted lines represent 0 .01 < P ≤ 0 .05 . The intensity of shading in the circles is proportional to the magnitude of the association. In the figure, “day” indicates the collection time of stool samples from sepsis patients after admission to the intensive care unit (ICU)(days 1 , 3 , and 7 after admission to the ICU). D-Lac: d-lactic acid; PCT: Procalcitonin; DAO: Diamine oxidase.
CONCLUSION
In this study, we report that sepsis patients in the ICU showed intestinal microecological disorders, lasting for at least 1 wk. Furthermore, intestinal microecological disorder was correlated with inflammation-related and intestinal barrier-related indexes in sepsis. However, this study is not without limitations. The sample size was small, and thus, it is necessary to carry out larger and multicenter studies to support these findings. Additional animal studies might be needed to clarify the related molecular mechanisms.
ARTICLE HIGHLIGHTS

ACKNOWLEDGEMENTS
We thank all the patients who participated in this study and the physicians in the Department of Critical Care Medicine, General Hospital, Ningxia Medical University.We also thank Shanghai Mobio Biomedical Technology Co. for technical support and to Ren HY for her help in writing the manuscript.
杂志排行

World Journal of Gastroenterology的其它文章
- Celiac Disease in Asia beyond the Middle East and Indian subcontinent: Epidemiological burden and diagnostic barriers
- Biomarkers in autoimmune pancreatitis and immunoglobulin G4-related disease
- Risk of hepatitis B virus reactivation in patients with autoimmune diseases undergoing non-tumor necrosis factor-targeted biologics
- Risk factors and prognostic value of acute severe lower gastrointestinal bleeding in Crohn’s disease
- Changes in the nutritional status of nine vitamins in patients with esophageal cancer during chemotherapy
- Gut microbiota dysbiosis in Chinese children with type 1 diabetes mellitus: An observational study