Risk of hepatitis B virus reactivation in patients with autoimmune diseases undergoing non-tumor necrosis factor-targeted biologics
2021-05-25ShintaroAkiyamaThomasCotterAtsushiSakuraba
Shintaro Akiyama, Thomas G Cotter, Atsushi Sakuraba
Abstract
Key Words: Hepatitis B virus; Autoimmune diseases; Biological therapy; Interleukin-23 ;Interleukin-17 ; Janus kinases
INTRODUCTION
Hepatitis B virus reactivation (HBVr) can occur in patients treated with immunosuppressive therapy and chemotherapy. In the current era of biologics, physicians need to understand the risk of HBVr in patients with autoimmune diseases undergoing anti-cytokine therapies.
The following three components are important for the development of HBVr: (1)The host immune response; (2 ) The covalently closed circular DNA of the viral genome of HBV (cccDNA); and (3 ) The use of immunosuppressive drugs[1 ]. HBV infection induces a series of innate[2 ] and adaptive[3 ] immune responses[1 ]. The host immune responses against hepatitis B virus (HBV) infection recruit adaptive cytotoxic T (Tc) cells to induce both cytolytic-dependent and -independent antiviral effects. In the cytolytic-independent effect, interferons (IFN) play an important role to suppress the HBV replication. To produce neutralizing antibodies to clear circulating HBV, B cells are also recruited to limit the viral spread of HBV (Figure 1 )[4 ]. However, even when clinical resolution of HBV infection is achieved, it does not mean complete elimination of HBV-DNA because cccDNA can persist in the nucleus of hepatocytes and it can be a source of HBVr when immunosuppressive medications are used.
Tumor necrosis factor (TNF)-α is a key cytokine not only in the pathogenesis of autoimmune diseases but also in the host immune reactions against HBV infection.TNF-α is synthesized by macrophages and T cells and induce the production of a variety of inflammatory cytokines, suppressing viral replication[5]. TNF-α is also necessary for the proliferation of HBV-specific Tc cells that are essential for suppression of HBV replication[6]. Hence, TNF-α inhibitors (e.g.,infliximab,adalimumab, and etanercept) can inhibit the anti-HBV immune response, leading to HBV replication[7]. Indeed, the pooled prevalence of HBVr in patients with autoimmune diseases undergoing TNF-α inhibitors was reported to be 4 .2 % (95 %CI:1 .4 %-8 .2 %)[8 ].
The signaling pathways involving interleukin (IL)-12 /23 , IL-17 , and Janus kinases(JAKs) have been highlighted as novel specific therapeutic targets for autoimmune diseases. A recent multicenter observational study for patients with psoriasis showed that HBVr was significantly more common among patients receiving anti-TNF-α therapies than IL-17 inhibitors[9 ]. However, there is still limited data in understanding the risk of HBVr in patients who are treated with biologics which inhibit such specific inflammatory pathways. In the present article, we aimed to review previous literatures which assessed the risk of HBVr in patients treated with non-TNF-targeted biologics and discuss how each medication can influence the development of HBVr.
DEFINITIONS OF HBV INFECTION AND REACTIVATION
The professional societies in the United States [American Association for the Study of Liver Diseases (AASLD); American Gastroenterological Association (AGA)], Europe[European Association for the Study of the Liver (EASL)] and Asia [Asian Pacific Association for the Study of the Liver (APASL)] have published guidelines to assist providers with HBVr management[10 -13 ]. In this review article, we divide patients into 2 risk groups which is consistent with the professional society guidelines[10 -13 ]when assessing practical management of HBVr.
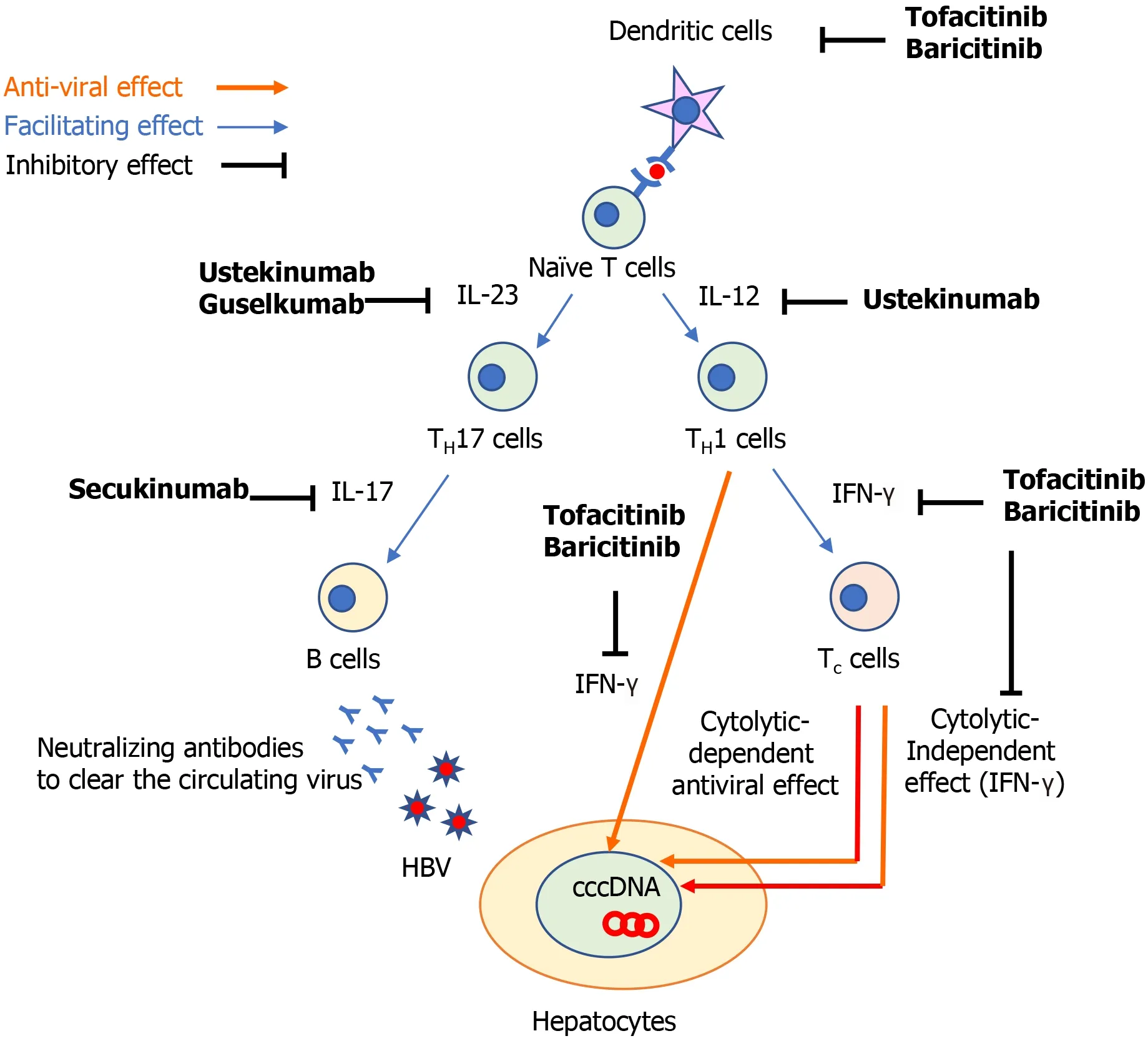
Figure 1 The possible immunological mechanism to explain how non-tumor necrosis factor-targeted biologics can induce the development of hepatitis B reactivation. cccDNA: Covalently closed circular DNA; HBV: Hepatitis B virus; IFN: Interferon; IL: Interleukin; TH17 cells: IL-17 producing T helper cells; TH1 cells: T helper 1 cells; Tc cells: Cytotoxic T cells.
Chronic HBV
Chronic HBV (CHB) [i.e.,Hepatitis B surface antigen (HBsAg)-positive and antibody to hepatitis B core antigen (anti-HBc)-positive] which includes patients with chronic active [serum HBV DNA ≥ 2000 IU/mL and normal or elevated serum alanine transaminase (ALT)] or inactive (serum HBV DNA < 2000 IU/mL and normal ALT) HBV infection.
Resolved HBV
Resolved HBV (i.e.,HBsAg-negative and anti-HBc-positive). Of note, there is insufficient evidence to support the use of anti-HBs titers as a decision aid when making a recommendation regarding prophylaxis[11 ].
There are subtle differences in the definition of HBVr among the professional society guidelines, however, the general concept is the same[10 -13 ]. In patients with CHB, HBVr is defined by a rise in HBV DNA above baseline. In patients with resolved HBV, HBVr is defined by either the appearance of HBV DNA in the blood or conversion to the HBsAg+ state (i.e.,seroreversion). The heterogeneity observed in HBVr definition is also reflected in the existing studies on HBVr. The majority of studies included the following parameters: (1 ) An acute rise in HBV-DNA levels compared with baseline; (2 ) Elevated levels of serum aminotransferases; and (3 )Seroreversion[1]. In this review article, we followed the criteria of HBVr described in each article.
THE MANAGEMENT OF PATIENTS WITH HBV INFECTION UNDERGOING IMMUNOSUPPRESSION
Risk stratification
Patients with CHB have an increased risk of HBVr when undergoing immunosuppressive therapy compared to patients with resolved HBV. For example, among patients treated with TNF-α inhibitors there is an estimated 5 -fold increased risk of HBVr in patients with CHB compared to patients with resolved HBV (15 .4 % vs 3 .0 %risk of HBVr)[8]. Further, another study showed that the pooled rate of HBVr without antiviral prophylaxis was 15 .6 % (95 %CI: 2 .3 -35 .7 ) in patients with CHB who were treated with TNF-α inhibitors. In patients with resolved HBV, the pooled rates of HBVr without antiviral prophylaxis in patients who were treated with TNF-α inhibitors and non-TNF-targeted biologics were 1 .4 % (95 %CI: 0 .5 %-2 .6 %) and 6 .1 %(95 %CI: 0 .0 %-16 .6 %), respectively[14 ]. Each of the 4 professional societal guidelines recommend testing HBV serology on all candidates for immunosuppressive therapy or chemotherapy to enable appropriate risk stratification (i.e.CHBvsresolved HBV)[10 -13 ].
Next, the degree of expected iatrogenic immunosuppression should be assessed.Hematopoietic stem cell transplant (HSCT) recipients and B cell–depleting therapies (e.g., rituximab) are high-potency regimens and confer the highest risk of HBVr[10 -13 ].The AGA guidelines ascertain that anthracyclines (e.g., doxorubicin) and moderate- to high-dose corticosteroids (CS) (i.e.,≥ 10 mg of daily prednisone or equivalent for ≥ 4 wk) confer higher risk than other immunosuppressants[11 ].
Therapeutic prophylaxis
CHB: In general, the professional societal guidelines recommend HBV prophylaxis,typically entecavir or tenofovir, for all candidates for immunosuppression who have CHB, apart from patients treated with traditional immunosuppressive agents (e.g.,thiopurines, methotrexate), intra-articular CSs, or oral CSs ≤ 1 wk[10 -13 ]. The AGA risk stratify this cohort of patients into moderate (1 %-10 %) and high risk (> 10 %)groups for HBVr[11 ]. Antiviral prophylaxis should be started before and continued after cessation of immunosuppression, generally 12 to 18 mo if high-potency therapies are used and 6 to 12 mo for other therapies[10 -13 ].
Resolved HBV: Guidelines largely agree that resolved HBV patients on high-potency immunosuppression (HSCT recipients and B cell–depleting therapies) should receive HBVr prophylaxis, with the AGA placing this group of patients in the high risk HBVr group (> 10 %)[10 -13 ].
For resolved HBV patients not on a high-potency regimen, the guidelines are more dissimilar. AGA recommend prophylaxis for resolved HBV patients at moderate risk(1 %-10 %) of HBVr, which include patients treated with TNF-α inhibitors, other cytokine or integrin inhibitors, tyrosine kinase inhibitors, moderate- or high-dose CSs for ≥ 4 wk and anthracycline derivatives. In contrast, AASLD, EASL and APASL recommend a pre-emptive therapy for this patient cohort, not prophylaxis, whereby serial lab monitoring (HBV DNA, HBsAg) is performed at 1 - to 3 -mo intervals on therapy and up to 12 mo after cessation of immunosuppression with on-demand antiviral therapy if needed[10 ,12 ,13 ]. Given that HBsAg seroreversion can lead to fatal acute hepatitis, antiviral therapy should be started immediately, independently of ALT level[11 ]. Of note, both EASL and APASL recommend treating resolved HBV patients similarly to HBsAg-positive patients if baseline serum HBV-DNA is positive[12 ,13 ].
AGA classify resolved HBV patients who are treated with traditional immunosuppressive agents (e.g., thiopurines, methotrexate), low-dose CSs ≥ 4 wk, intra-articular CSs, or any dose of oral CSs for ≤ 1 wk, as low-risk (< 1 %) for HBVr and do not recommend prophylaxis, similar to the other society guidelines[10 -13 ].
The risk of HBVr in patients who are treated with non-TNF-targeted biologics
Given the paucity of data on the HBVr risk among patients treated with non-TNFtargeted biologics, we reviewed the existing literature on the risk of HBVr in patients with autoimmune diseases who received non-TNF-targeted biologics and summarized the findings in Tables 1 -3 . A majority of articles focused on patients with CHB or resolved infection. According to the AGA guideline, CHB and resolved HBV patients treated with non-TNF-targeted therapies are categorized into the moderate-risk HBVr group and therapeutic prophylaxis is recommended. However, AASLD, EASL and APASL recommend serial monitoring among resolved HBV (if HBV DNA is negative)with pre-emptive prophylaxis if HBVr is observed. Therefore, it is important to determine the precise HBVr with non-TNF-targeted biologics to ascertain if a strategy of monitoring/pre-emptive may be too lax, and perhaps a uniform strategy of prophylaxis may be more optimal as recommended by the AGA. Given that patients with resolved infection should be treated similarly to those with CHB patients if their serum HBV-DNA tests are positive at baseline[12 ,13 ], we present their baseline HBVDNA in Tables 1 -3 .
IL-12 /23 INHIBITORS
Mechanism of HBV reactivation
The cytokine IL-12 contributes to the differentiation of naïve T cells to T helper 1 (TH1 )cells and IL-23 maintains and expand IL-17 producing T helper (TH17 ) cells (Figure 1 )[15 ]. These two cytokines play a central role to regulate T cell-mediated immune responses, which are dysregulated in various autoimmune diseases including psoriasis and Crohn’s disease (CD)[15 ,16 ]. The clinical benefit of IL-12 and IL-23 inhibition has been demonstrated in psoriasis, CD, and ulcerative colitis by ustekinumab[17 -19 ],which is an antibody against p40 , the common subunit of IL-12 and IL-23 . IL-12 plays an important role in achieving sustained control of HBV replication. IL-12 can promote cell-mediated immunity by facilitating the production of IFN-γ production by TH1 cells, resulting in the inhibition of HBV replication[20 ,21 ] and the induction of antiviral effects of HBV-specific Tc cells[22 ,23 ]. Indeed, patients with CHB who were treated with recombinant human IL-12 exhibited a high proportion of HBV clearance in a dose-dependent manner[24 ] and the addition of IL-12 to lamivudine enhanced T cell reactivity to HBV and IFN-γ production[25 ]. Furthermore, patients with CHB responding to IFN-α treatment were shown to have higher IL-12 and IFN-γ expression levels during the treatment[26 ]. These findings suggest that ustekinumab might theoretically increase the risk of HBVr.
Clinical studies in patients with autoimmune diseases
Several studies have assessed the risk of HBVr in patients treated with ustekinumab(Table 1 ). A retrospective study showed that no HBVr occurred among 2 patients with CHB taking antiviral prophylaxis after starting ustekinumab, whereas, 2 patients(25 %) developed HBVr without hepatitis among 8 patients with CHB without antiviral prophylaxis[27 ]. Another study demonstrated a 29 % rate of HBVr after ustekinumab initiation without antiviral prophylaxis in patients with CHB[28 ]. Given that patients with CHB have a high risk of HBVr without antiviral prophylaxis, these patients require antiviral prophylaxis and appropriate monitoring for HBV-DNA and serology tests after initiating ustekinumab treatment.
A retrospective study on 44 patients with resolved HBV who initiated ustekinumab without antiviral prophylaxis found that 1 patient (2 .3 %) developed HBVr complicated with mild hepatitis[27 ]. This patient discontinued concurrent methotrexate when reactivation occurred and HBV-DNA became undetectable without antiviral therapy in 6 mo. In another study involving 7 patients with resolved HBV on ustekinuamb, 1 patient (14 .3 %) developed HBVr. This patient was not on antiviral prophylaxis and started entecavir for treatment of HBVr[5]. These data suggest that there is a certain risk of HBVr in patients with resolved HBV even without detectable HBV-DNA at baseline after starting ustekinumab, suggesting that these patients might need antiviral prophylaxis as is the AGA guidelines preferred option. While the guidelines of AASLD, EASL, APASL recommend pre-emptive therapy if HBV DNA is negative,further studies are warranted in order to understand if these patients require antiviral prophylaxis.
IL-23 INHIBITORS
Mechanism of HBV reactivation
IL-23 -specific antagonists, such as tildrakizumab[29 ,30 ], risankizumab[31 ,32 ], guselkumab[33 ,34 ], and brazikumab[35 ], have been shown to be effective for psoriasis and CD.These medications bind to the p19 subunit on IL-23 and inhibit its interaction with the IL-23 receptors[36 ]. The potential mechanism of HBVr in patients treated with IL-23 inhibitors is still unclear. Previous studies found that TH17 cells, which are expanded by IL-23 (Figure 1 ), increase with the severity of liver damage in patients with CHB[37 -39 ]. An observational, clinical-controlled study also demonstrated that the expression levels of IL-23 and IL-17 were associated with increased possibilities of hepatitis B e-antigen (HBeAg) clearance and HBsAg decline in patients with HBeAgpositive CHB during pegylated IFN therapy[40 ]. This study also found that high serum IL-23 Levels can predict the response to IFN therapy in patients with HBeAgpositive CHB[40 ]. Given that TH17 cells promote the differentiation and function of B cells[41 ,42 ], IL-23 might activate the humoral immune response against circulating HBV and play a role to facilitate HBV clearance by IFN therapy (Figure 1). Although this hypothesis suggest that IL-23 inhibitors may abrogate the HBV clearance, it still remains to be elucidated whether these medications contribute to the development of HBVr.
Clinical studies in patients with autoimmune diseases
The data regarding the safety of IL-23 inhibitors in patients with HBV infection is limited. A case report showed that a patient with resolved HBV infection did not develop HBVr 1 year after starting guselkumab (Table 1 )[43 ]. There have been no reported studies focusing on the risk of HBVr in patients treated with other IL-23 inhibitors.
IL-17 INHIBITORS
Mechanism of HBV reactivation
IL-17 is a major effector cytokine of TH17 cells and mediate host defense mechanisms[44 ]. Inhibition of IL-17 with secukinumab, ixekizumab, and brodalumab have demonstrated clinical benefits in patients with psoriasis[45 -47 ], psoriatic arthritis[48 ], and ankylosing spondylitis[49 ]. TH17 /IL-17 axis is involved in the process of fibrogenesis and increases the expression of proinflammatory cytokines, promoting the recruitment of inflammatory cells in patients with CHB[50 ]. A previous study showed that Th17 cells were significantly increased in patients with CHB, as well as the expression level of IL-17 [51 ]. They also demonstrated that the suppression of viral replication induced by IFN-α resulted in a decrease in TH17 cells and IL-17 expression,suggesting that TH17 cells might play an important role during IFN-α treatment to eliminate HBV[51 ]. As we described above, TH17 cells also facilitate B cells[41 ,42 ] and would enhance the humoral response to clear circulating HBV (Figure 1). These findings implicate that TH17 /IL-17 axis might be associated with HBV clearance and its inhibition may increase the risk of HBVr.
Clinical studies in patients with autoimmune diseases
A prospective multicenter study on 22 patients with CHB with no antiviral prophylaxis after starting secukinumab showed that 6 patients (27 .3 %) developed HBVr (Table 2 )[52 ]. Three patients with HBVr started antiviral treatments and their viral loads decreased rapidly within 3 mo. The remaining three patients with HBVr were followed without antiviral drugs and their viral loads remained low without acute hepatitis. Notably, none of the 3 patients with CHB who received antiviral prophylaxis developed HBVr[52 ]. Hence, this study reinforced the importance of antiviral prophylaxis in patients with CHB starting treatment with IL-17 inhibitors.This study also included 24 patients with resolved HBV who did not receive antiviral prophylaxis and identified one patient (4 .2 %) with a positive viral load at baseline who developed HBVr without acute hepatitis[52 ]. This study re-affirmed the EASL and APASL guidelines which recommend antiviral prophylaxis in patients with resolved HBV if their baseline viral loads are positive.
A case report on a patient with CHB treated with ixekizumab and entecavir simultaneously did not develop HBVr after 18 mo of treatment[53 ]. Another report showed that a patient with resolved HBV did not experience HBVr during follow-up(Table 2 )[54 ]. Given that data regarding the risk of HBVr in patients treated with ixekizumab or brodalumab are still limited, further studies with larger sample sizes are warranted.
JAK INHIBITORS
Mechanism of HBV reactivation
JAKs bind to type I and II cytokine receptors and transmit extracellular cytokine signals to activate various signal transducers and activators of transcription, which drive the proinflammatory machinery of the cellular immune response[55 ]. The clinical benefit of JAK inhibitors has been demonstrated in patients with rheumatoid arthritis[56 ,57 ], psoriatic arthritis[58 ,59 ], and ulcerative colitis[60 ,61 ]. Important signaling pathways in host-defense include innate antiviral responsesviaIFN-α/β mediated by JAK1 -tyrosine kinase 2 complexes, and IFN-γ mediated by JAK1 -JAK2 complexes[55 ]. Hence, JAK inhibitors might counteract the suppressive effects of IFN on viral replication[62 ,63 ]. Further, dendritic cells and effective T cell lineages including THcells and Tc cells play important roles to defense against HBV-infection(Figure 1 )[64 ]. A previous study demonstrated that a JAK inhibitor can block the differentiation and function of dendritic cells, leading to impaired T cell activation(Figure 1 )[65 ]. Thus, it was suggested that JAK inhibitors might negatively interact with the defense mechanism against HBV infection. Further studies investigating how JAK inhibitors influence the development of HBVr are warranted.
Clinical studies in patients with autoimmune diseases
A retrospective cohort study including 6 patients with CHB showed that 2 out of 4 patients (50 %) without antiviral prophylaxis developed HBVr after starting tofacitinib.One patient had an elevated ALT level and started entecavir, resulting in declines in HBV-DNA and ALT levels. Another patient started entecavir and did not develop acute hepatitis. Both patients continued tofacitinib after the development of HBVr.Meanwhile, 2 patients with CHB who received antiviral prophylaxis did not develop HBVr after initiating tofacitinib. Further, in this study, none of 75 patients with resolved HBV received antiviral prophylaxis and no HBVr was observed in this group[66 ]. Another study also demonstrated that patients with resolved HBV did not develop HBVr after starting tofacitinib (Table 3 )[67 ].
A study assessing data which was integrated from four phase 3 trials of baricitinib in patients showed that, among 215 patients with resolved HBV, 8 patients (3 .7 %) had a single quantifiable result of HBV-DNA viral load (HBV-DNA level ≥ 29 IU/mL) after initiating baricitinib. Among these 8 patients, 4 patients met the definition of HBVr(HBV-DNA ≥ 100 IU/mL), but no patients developed hepatitis. HBV-DNA at baseline was assessed in 6 patients and all examined patients did not have detectable HBVDNA level. Antiviral therapy was not used in 5 of 8 patients[62 ].
All these findings suggest that patients with CHB should receive antiviral prophylaxis when they start JAK inhibitors. As for patients with resolved HBV infection, given that HBVr was occasionally reported even if their HBV-DNA levels were not detected at baseline, an appropriate consultation with hepatologists is necessary. There has been limited data regarding the risk of HBVr in patients with autoimmune diseases who are treated with other JAK inhibitors (e.g.,upadacitinib,filgotinib, peficitinib).
CONCLUSION
In summary, considering antiviral prophylaxis with an appropriate risk stratification is necessary when we start non-TNF-targeted biologics for patients with autoimmune diseases. The frequencies of HBVr without antiviral prophylaxis in patients with CHB on IL-12 /23 , IL-17 , and JAK inhibitors are up to 29 %, 27 %, and 50 %, respectively. A meta-analysis demonstrated that the pooled rate of HBVr without antiviral prophylaxis was 15 .6 % (95 %CI: 2 .3 -35 .7 ) in patients with CHB who were treated with TNF-α inhibitors, suggesting that non-TNF-targeted biologics, particularly JAK inhibitors, may have a higher risk of HBVr compared with TNF-α inhibitors. Given that no patients who received antiviral prophylaxis developed HBVr, HBVr is preventable with antiviral therapy in patients with CHB on non-TNF-targeted biologics. As all of professional societies recommended in their guidelines, patients with CHB should receive antiviral prophylaxis when they start non-TNF-targeted biologics. In patients with resolved HBV, the rates of HBVr without antiviral prophylaxis in patients on IL-12 /23 , IL-17 , and JAK inhibitors are up to 2 .3 %, 4 .2 %,and 0%, respectively. The meta-analysis showed that the pooled rates of HBVr without antiviral prophylaxis in patients who were treated with TNF-α inhibitors and non-TNF-targeted biologics were 1 .4 % (95 %CI: 0 .5 %-2 .6 %) and 6 .1 % (95 %CI: 0 .0 %-16 .6 %),respectively[14 ]. These data supported that the risk of HBVr in patients treated with non-TNF-targeted biologics might be higher than that in patients with TNF-α inhibitors even if their HBV status is resolved HBV. According to the AGA guideline,patients with resolved HBV who are treated with non-TNF-targeted biologics are categorized into the moderate risk group and antiviral prophylaxis are recommended for this patient cohort[11 ]. However, as stated previously, AASLD, EASL and APASL recommend the pre-emptive therapeutic strategy for this cohort, although APASL and EASL do include the caveat of potentially using HBV DNA assessment to aid decisionmaking[10 ,12 ,13 ]. Given the higher risk of HBVr with non-TNF-targeted biologics compared with TNF-α inhibitors, antiviral prophylaxis may be a favorable strategy rather than the pre-emptive strategy to prevent HBVr in patients with resolved HBV.Large-scale studies are needed to ascertain the differential risk of HBVr between patients with TNF-α inhibitors and non-TNF-targeted biologics and to stratify the risk of HBVr by the type of non-TNF-targeted biologics. While HBsAg seroreversion can lead to fatal acute hepatitis, a consultation with hepatologists or infectious disease specialists is recommended.
杂志排行

World Journal of Gastroenterology的其它文章
- Celiac Disease in Asia beyond the Middle East and Indian subcontinent: Epidemiological burden and diagnostic barriers
- Biomarkers in autoimmune pancreatitis and immunoglobulin G4-related disease
- Risk factors and prognostic value of acute severe lower gastrointestinal bleeding in Crohn’s disease
- Changes in the nutritional status of nine vitamins in patients with esophageal cancer during chemotherapy
- Effects of sepsis and its treatment measures on intestinal flora structure in critical care patients
- Gut microbiota dysbiosis in Chinese children with type 1 diabetes mellitus: An observational study