The transcriptional landscape of cultivated strawberry(Fragaria×ananassa) and its diploid ancestor (Fragaria vesca)during fruit development
2021-05-23LlYongpingLlUTianjiaLUOHuifengLlUShengcai
Ll Yong-ping ,LlU Tian-jia,LUO Hui-feng,LlU Sheng-cai
1 Institute of Horticultural Biotechnology,Fujian Agriculture and Forestry University,Fuzhou 350002,P.R.China
2 School of Life Sciences and State Key Laboratory of Agrobiotechnology,The Chinese University of Hong Kong,Hong Kong 999077,P.R.China
3 Key Laboratory of Horticultural Plant Biology (Ministry of Education),College of Horticulture and Forestry Sciences,Huazhong Agricultural University,Wuhan 999077,P.R.China
4 Institute of Horticulture,Hangzhou Academy of Agricultural Sciences,Hangzhou 310024,P.R.China
Abstract Cultivated strawberry (Fragaria×ananassa) originated from four diploid ancestors:F.vesca,F.viridis,F.iinumae and F.nipponica. Among them,F.vesca is the dominant subgenome for cultivated strawberry. It is not well understood how differences in gene expression between diploid and octoploid strawberry contribute to differences during fruit development.In this study,we used comprehensive transcriptomic analyses of F.vesca and F.×ananassa to investigate gene expression at the different stages of fruit development. In total,we obtained 3 508 (turning stage) and 3 958 (red stage) differentially expressed genes with pairwise comparisons between diploid and octoploid. The genes involved in flavonoid biosynthesis were almost upregulated in the turning stages of octoploid,and we also discovered a ripe fruit-specific module associated with several flavonoid biosynthetic genes,including FveMYB10,FveMYB9/11,and FveRAP,using weighted gene coexpression network analysis (WGCNA). Furthermore,we identified the species-specific regulated networks in the octoploid and diploid fruit. Notably,we found that the WAK and F-box genes were enriched in the octoploid and diploid fruits,respectively. This study elucidates new findings on flavonoid biosynthesis and fruit size of strawberry with important implications for future molecular breeding in cultivated strawberry.
Keywords:strawberry,Fragaria vesca,Fragaria×ananassa,RNA-Seq,fruit,development,anthocyanin
1.lntroduction
Whole-genome duplication (WGD) or polyploidy is an important evolutionary force in seed plants and angiosperms(Jiaoet al.2011),which probably contributed to their varied and novel phenotypes. Polyploids are generally grouped into two types,autopolyploids and allopolyploids,which are involved in single or different diploid progenitors,respectively (Stuparet al.2007). Allopolyploids originate primarily from interspecific hybridization and are composed of a complicated genetic background;therefore,such species contain a range of genetic alterations (Yanget al.2014;Zhanget al.2015). The composition of different subgenomes can significantly repress the phenotype of allopolyploids and commonly reflects evolutionary and domestication influences (Comai 2005). Numerous crop species are allopolyploids,including sorghum,cotton,potato,wheat and sugarcane. Cultivated strawberry(Fragraia×ananassa)is also an allopolyploid (octoploid)species,whose fruit exhibit a large size,disease resistance,intense color,and varied aromas and flavors compared with diploid varieties (Hancock and Bringhurst 1981;Rousseau-Gueutinet al.2008,2009).
Transcriptional remodeling in polyploids is caused by the interactions of diverged parental genomes that are reunited in the allopolyploid and genome duplications. It is likely to contribute to heterosis and exhibit variation in adaptation to new conditions (Comai 2005). Polyploidy changes the progress of meiosis and mitosis and the relationships between cellular components through epigenetic changes that modify gene expression (Comai 2005;Chen and Ni 2006). Furthermore,gene dosage balance andtransand/orcis-regulatory networks also affect the amount of gene expression. During the process of polyploidization,the splicing sites could be altered with changes in the components,abundance and activity of splicing factors,which could affect the absence and presence of alternative splicing (AS) in polyploids (Zhouet al.2011). The AS could create new isoforms,and therefore change the proteins and possibly also regulate the level of expression of transcripts through triggering nonsense-mediated decay (NMD)pathway degradation (Yooet al.2014). The AS events of diploid and octoploid strawberry have been identified (Liet al.2017;Yuanet al.2019),but the difference between them remains unclear,particularly during the process of fruit development.
Strawberry fruit is known to be a source of polyphenol compounds,particularly flavonoids,a large family of secondary metabolites. The flavonoids found at the highest levels include proanthocyanidins (PAs),followed by anthocyanins,flavonols and other phenolics (Almeidaet al.2007;Carboneet al.2009). Fruit color is one of the breeding objectives of substantial interest in strawberry.The anthocyanin biosynthetic genes and transport genes have been identified in strawberry (Almeidaet al.2007;Pilletet al.2015). The down-regulation of strawberryFaGT1can redirect flavonoid biosynthesis in ripening fruit(Griesseret al.2008).FaMYB10plays a general regulatory role in anthocyanin biosynthesis during strawberry ripening(Medina-Pucheet al.2013). Furthermore,there are some critical genes involved in anthocyanin biosynthesis or transport,such asFaMYB9/FaMYB10(Schaartet al.2013)and glutathioneS-transferase (GST) (Luoet al.2018). The availability of genomic and transcriptomic studies provides an opportunity to reveal the flavonoid biosynthetic pathway.However,no large-scale omics analyses have been performed to dissect the molecular mechanism of flavonoid biosynthesis in strawberry. Flavonoids have potential health benefits,and anthocyanins may be involved in plant growth and development,stress responses and gene expression.
The plant cell wall is primarily composed of cellulose,hemicellulose,pectin and proteins. The wall is thought to be involved in modulating plant development (Kohorn 2000).Cell expansion (Cosgrove 1997) and pathogen infection(Hammond-Kosack and Jones 1996) can alter biosynthesis,cell wall components and downstream cytoplasmic events,such as systemic acquired resistance. These events show that communication between the cytoplasm and the cell wall is of substantial importance. Wall-associated kinases(WAKs) are one of the four classes of proteins that physically link the ECM to the plasma membrane and may promote communication between the two compartments. WAKs belong to the group of receptor-like kinases,which have a cytoplasmic protein kinase domain and link to the pectin fraction of the plant cell wall. WAKs proteins regulate diverse biological processes,and their main function is their involvement in the pathogen response and plant cell expansion. Numerous environmental stimuli can activate their expression.
Fragaria vescais a wild diploid strawberry and subgenomic donor of the octoploid genome of cultivated strawberry (F.×ananassa) (Edgeret al.2019). However,the underlying mechanism and ultimate consequences of subgenome dominance are still mostly unknown. In this study,we successfully collected the spatial and temporal transcriptome data of diploid and octoploid strawberry fruit tissues at different stages of fruit ripening. Global transcriptome analysis showed that octoploid strawberry exhibited different transcriptome profiles in comparison with the diploid strawberry. It is helpful for breeding to reveal the transcriptional network and to identify potential genes and functional classes that are involved in strawberry fruit ripening. As a whole,this study contributes new information on the flavonoid biosynthetic processes and fruit size of strawberry,with important implications for future molecular breeding in the cultivated strawberry.
2.Materials and methods
2.1.Plant materials
The receptacle was collected from the diploidF.vesca‘YW5AF7’ (Slovinet al.2009) and a widely grown cultivar Sweet Charlie,which were germinated and grown in a greenhouse under a natural photoperiod. The receptacles(fruits) of both varieties were collected in the morning in late March. Equal amounts of the receptacle tissues of diploid and octoploid from each stage were pooled. Each sample was collected from two or three plants. The receptacles with achenes removed were quickly frozen in liquid nitrogen and stored at–80°C for RNA isolation.
2.2.lllumina RNA-sequencing of pooled receptacle tissue from YW5AF7 and Sweet Charlie
Equal amounts of receptacle tissues at small green to ripe stages were pooled for each variety (F.vesca‘YW5AF7’andF.×ananassa‘Sweet Charlie’). Total RNA was extracted and evaluated as described above (Liet al.2017). A total of 6–16 μg of RNA per sample was sent to the Beijing Genomics Institute (Wuhan,China) for strand-specific library construction and sequencing on an Illumina Hi-Seq 2500 platform (San Diego,CA,USA). A total of 89 and 142 million 125 bp paired-end reads were generated for YW5AF7 and Sweet Charlie,respectively. The RNA-Seq data were uploaded to the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/sra/) under the accession numbers SRX1756111 and SRX1756112,respectively.
2.3.Description of previous diploid and octoploid fruit datasets
We collected a total of 80 Illumina-based datasets representing 36 different fruit tissues of diploid and octoploid fruit at varying developmental stages,including seven development stages for diploid receptacles (cortex and pith) and other fruit tissues (wall,style,embryo,ovule and ghost) (Kanget al.2013;Hawkinset al.2017),as well as the four developmental stages (green,white,turning and red) for the octoploid achene and receptacle (Sánchez-Sevillaet al.2017). Furthermore,we collected the diploid receptacle samples at two different stages:the turning and red stages (Guet al.2019).
2.4.Read mapping and transcript assembly
Raw data were quality-and adaptor-trimmed using FastXToolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Next,clean reads were aligned toF.vescaGenome V4.0 (Edgeret al.2018) downloaded from GDR using the program STAR(Dobinet al.2013) with default parameters. Stringtie (Perteaet al.2015) was used to individually assemble transcripts based on a reference-guided assembly strategy. Putative transcripts for each sample were merged into a unique set of transcripts using TACO software (Niknafset al.2017). The merged transcripts file was then compared with the V4.0.a2(Liet al.2019) annotation GTF file using Cuffcompare(Trapnellet al.2012).
2.5.Differential expression analysis
The gene-level count matrices for annotation genes were generated using FeatureCount (Liaoet al.2013). The median-of-ratios method was used to normalize the data for RNA composition and sequencing depth using the DESeq2 package (Loveet al.2014). DESeq2 Software was used to identify differentially expressed genes (DEGs) based on the negative binomial generalized linear model. The DEGs were determined based on an absolute log2-fold change value of>4 andP-value adjusted (padj)<0.001.
2.6.Functional classification of genes based on the MapMan pathway and GO enrichment
MapMan Software (Thimmet al.2004) was utilized to facilitate the assignment of DEGs into functional categories(bins). MapMan bins of theF.vescaV4 genome annotation were assigned using the Mercator pipeline (https://mapman.gabipd.org/app/mercator). GO terms for the full set of genes in v4.0.a2 annotation were assigned using Blast2GO(Conesa and Götz 2008),and significantly enriched GO terms were identified based on a hypergeometric test with a false discovery rate (FDR)<0.05.
2.7.AS event and differential AS gene detection
To classify the AS events,rMATS software (Shenet al.2012) was utilized with the transcript GTF files assembled from diploid and octoploid data. Five major types of AS events,namely retained intron (RI),alternative 3´ splice site (A3SS),alternative 5´ splice site (A5SS),skipped exon(SE) and mutually exclusive exons (MXE),were counted for each sample separately. rMATS was also used to detect the differentially spliced events between the diploid and octoploid using the aligned BAM files as input with default parameters. The merged transcripts GTF file generated by the Cuffcompare software was used as the reference. An AS event between diploid and octoploid with an FDR<0.05 and“Exon Inclusion Level Difference”> 0.1 was defined as a differentially spliced event.
2.8.Validation of differential AS events by RT-PCR
Total RNA was isolated from the pooled receptacles of diploid and octoploid fruit using a Plant Total RNA Isolation Kit (No.SK8631,Sangon Biotech,Shanghai,China). The cDNA was synthesized using a PrimeScript RT reagent kit with gDNA Eraser (TaKaRa,Shiga,Japan). cDNA diluted five-fold served as the template. KOD DNA polymerase(Cat# F0934K,TOYOBO Bio-Tech,Osaka,Japan) was used for PCR amplification. Finally,the PCR products were visualized by staining on an agarose gel.
2.9.Weighted gene co-expression network analysis
A co-expression network analysis was performed using the WGCNA package in R (Langfelder and Horvath 2008). The raw counts for 26 194 genes (transcripts per million (TPM)>2 in at least one tissue) were normalized to homoscedastic expression values using the varianceStabilizingTransformation function in the DESeq2 package (Loveet al.2014). A signed hybrid network was constructed using the automatic network construction function blockwiseModules that calculated the topographical overlap matrices (TOM) with default settings,with the exceptions that the power=20,TOMType=“signed”,minModuleSize=30 and mergeCutHeight=0.25. To determine the associations of modules with the tissuespecific expression for each tissue,the correlation (cor ())function was used for the correlations between modules and tissues. A summary profile (module eigengene,ME)was calculated for each module using Principal Component Analysis (PCA). The most highly connected genes are central hub genes. Finally,the network was visualized using Cytoscape_v3.6.1 (Smootet al.2010).
3.Results
3.1.Comparative analysis of transcriptome sequencing data between the diploid and octoploid strawberry
YW5AF7 (F.vesca) is a diploid wild strawberry primarily used for basic research,whereasF.×ananassais an octoploid commercial variety. The fruit flesh ofF.×ananassais quite different from YW5AF7 in terms of size,color,aroma and texture (Fig.1-A). To understand the effect of ploidy on global gene expression,we collected a total of 86 RNA-Seq libraries generated from 38 different tissue types,including four fruit development stages (green,white,turning and red)for the octoploid achene and receptacle,as well as eight development stages for the diploid receptacle (cortex and pith) and other fruit tissues (wall,style,embryo,ovule and ghost). A total of 2.6 billion RNA-Seq reads were generated from the diploid and octoploid receptacles. SinceF.vescais a near-complete genome with well annotated genes that contribute to one set of the subgenomes ofF.×ananassa,its genome was used as a reference in our analyses. The clean reads were mapped to theFragaria vescaV4 genome(Edgeret al.2018) using annotation V4.0.a2 (Liet al.2019)with mapping ratios of 95% for diploid samples and 85%for octoploid samples,respectively (Appendix A). The transcriptome profile from the octoploid samples exhibited a large distance from those of the diploid strawberry. The clustering results indicated that the polyploid effect could largely explain the variation among samples. Overall,our results suggested that the quantity and quality of RNA-Seq data were suitable for downstream comparative analyses.
3.2.Transcriptome is altered between diploid and octoploid fruit during ripening
Turning and red are two representative stages of strawberry fruit ripening. At the turning stage,the diploid and octoploid fruit have red skin. At the red stage,they have similarly colored skin and differentially colored flesh. The Pink and Red samples (Appendix A) were the turning and red stages in the diploid (Guet al.2019),consistent with the turning receptacle and red receptacle in octoploid,respectively.We collected the RNA-Seq datasets of the turning and red stages for both octoploid and diploid strawberry fruit to gain a genome-wide view of the gene expression at these two stages (Fig.1-A). Pairwise comparisons (Pinkvs.turning receptacle,Redvs.red receptacle) were then performed to identify differentially expressed genes (DEGs)using DESeq2. We obtained a total of 3 508 (turning) and 3 958 (red) DEGs between diploid and octoploid. At the turning stage,pairwise comparisons of octoploid and diploid showed that 1 806 genes were significantly upregulated,while 1 702 were significantly downregulated (|fold change|>4,padj≤0.001) (Appendix B). At the red stage,1 530 and 2 428 genes were significantly upregulated and downregulated,respectively,in samples of the red stages of octoploid compared with those of the diploid. Further analysis indicated that only one gene (FvH4_7g14680) was upregulated in the two sets of pairwise comparisons. It is an AtRD22 homologous gene,which is involved in abscisic acid (ABA)-mediated stress responses (Matuset al.2014).In addition,no gene was commonly downregulated.
Furthermore,MapMan bins were used to identify specific categories of genes or pathways that were enriched in differentially expressed genes. As shown in Fig.1-C,the secondary metabolism pathway analysis suggested that the genes involved in flavonoids and chalcones were almost upregulated in the turning stage and downregulated in the red stage of octoploid. All the results obtained are consistent with the upregulation of flavonoid and anthocyanin metabolism inF.ananassaflesh,while an opposite trend was observed inF.vesca. A similar phenomenon has been reported in two octoploid strawberries:Xiaobai (whitefleshed) and Benihoppe (red-fleshed) (Linet al.2018).

Fig.1 Summary of differentially expressed genes (DEGs) between diploid and octoploid strawberry fruits in the turning and red stages. A,photographs of diploid and octoploid fruit tissues in the turning and red stages. B,numbers of DEGs detected between diploid and octoploid in the turning and red stages. C,secondary metabolism overview corresponding to the DEGs in the turning and red stages. The color key indicates the log2 -fold change between diploid and octoploid. Red denotes upregulation and green downregulation.
3.3.AS events between diploid and octoploid receptacles
To investigate the impacts of ploidy on AS and the contributions of AS to fruit quality,fruit (receptacle)transcriptomes fromF.×ananassa‘Sweet Charlie’ andF.vescawere obtained using Illumina RNA-Seq. After comparing the isoforms in diploid and octoploid,a larger number of isoforms was uniquely identified in the octoploid(28 662 octoploid versus 17 759 diploid) despite the fact that there was adequate sequencing depth in our data(Fig.2-A). The AS events in diploid and octoploid were further classified into different types,including RI,A3SS,A5SS,SE and MXE using the rMATS tools. Consistent with the isoform numbers,more AS events were also identified in the octoploid samples (Fig.2-B). RI was the most frequent AS type for both varieties,followed by SE,A3SS,A5SS and MXE,similar to other plant species (Campbellet al.2006;Sun and Xiao 2015;Wanget al.2016). This result indicated that the global AS regulation in octoploid is similar to that in the diploid.
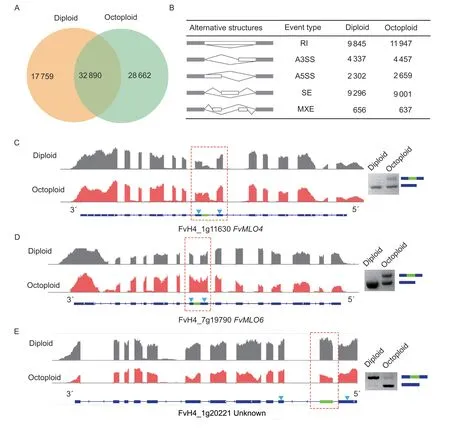
Fig.2 Large-scale identification of alternative splicing in diploid and octoploid using RNA-Seq. A,a Venn diagram showing the common and unique transcripts from diploid and octoploid. B,statistics of different AS events obtained from diploid and octoploid fruit. C,D,and E,PCR validation of the differential alternative splicing events of FvMLO4,FvMLO6 and FvH4_1g20221 between diploid and octoploid,respectively. For each gene,read coverage graphs viewed in IGV are shown with a red dotted square indicating the differentially spliced regions between the two samples. The gene model is shown on the bottom with blue arrowheads indicating the positions of primers used for RT-PCR validation. The agarose gel image shows the bands of two transcript variants amplified from the same RNA samples for sequencing with gene models to the right. AS,alternative splicing;IGV,Integrative Genomics Viewer.
It is difficult to determine the level of expression of each isoform obtained by Illumina RNA-Seq. AS events were compared individually between the diploid and octoploid using rMATS to identify potential AS genes that regulated fruit quality (Shenet al.2012). As shown in Appendix C,1 779 and 2 284 AS events,respectively,were more abundant in octoploid and diploid (FDR<0.05,absolute“Exon Inclusion Level Difference”>10%). For example,FvH4_1g11630,designatedFvMLO4,encoded a homolog of the barley mildew resistance locus o (MLO) protein(Pessinaet al.2014). The gene has one more 3´ splicing site in the eighth exon with a 41.9% higher exon inclusion level in octoploid that results in a premature stop codon and was validated by RT-PCR (Fig.2-C). Interestingly,another MLO protein-encoding gene FvH4_7g19790,designatedFvMLO6(Pessinaet al.2014),also possesses a differential splicing event with a retained intron (Fig.2-D). This similarity suggests that the MLO gene family is susceptible to the AS regulation and potentially contributes to the powdery mildew resistance of Sweet Charlie. In addition,FvH4_1g20221,encoding an unknown protein,also exists inMalus domesticaandPrunus persica,and its isoform skipped the entire second exon specifically in the octoploid samples leading to the deletion of 71 amino acids (Fig.2-E).In summary,although differences in AS events were found in the receptacles of diploid and octoploid strawberries,the characteristics of regulation of AS are similar.
3.4.Coexpression network analysis with WGCNA
The dynamic expression patterns of genes reflect their roles during fruit development. RNA-Seq data for 36 different tissues/stages were generated from diploid (28 samples)and octoploid (8 samples). To comprehensively understand the gene regulatory networks of the two varieties and identify specific genes associated with polyploids and the flavonoid biosynthetic pathway during fruit ripening,we performed the WGCNA. After filtering the low expression genes (TPM>2 in at least one tissue),26 194 genes were retained for WGCNA. The cluster dendrogram revealed the correlation of transcriptomes of different tissues in diploid and octoploid samples (Fig.3-A) after using the“hclust”function matrix.Different stages of the same tissue from diploid and octoploid were found to be similar and could be clustered together.
Coexpression networks were constructed on the basis of pairwise correlations of gene expression of all the sample tissues. Using standard parameters,the genes could be assigned to 26 distinct modules (labeled by different colors)(Fig.3-B). Consistent with cluster analysis,the tissues were a major driver of module identity according to eigengene tissue-specific expression profiles (Fig.3-C,Appendix D),which allow for visualization of the cluster-tissue association.For example,genes in the salmon color module are the most highly expressed in style 1. The brown color module correlates with the octoploid achenes. The large number of tissues and stages of the diploid and octoploid enabled the development of fruit coexpression networks.
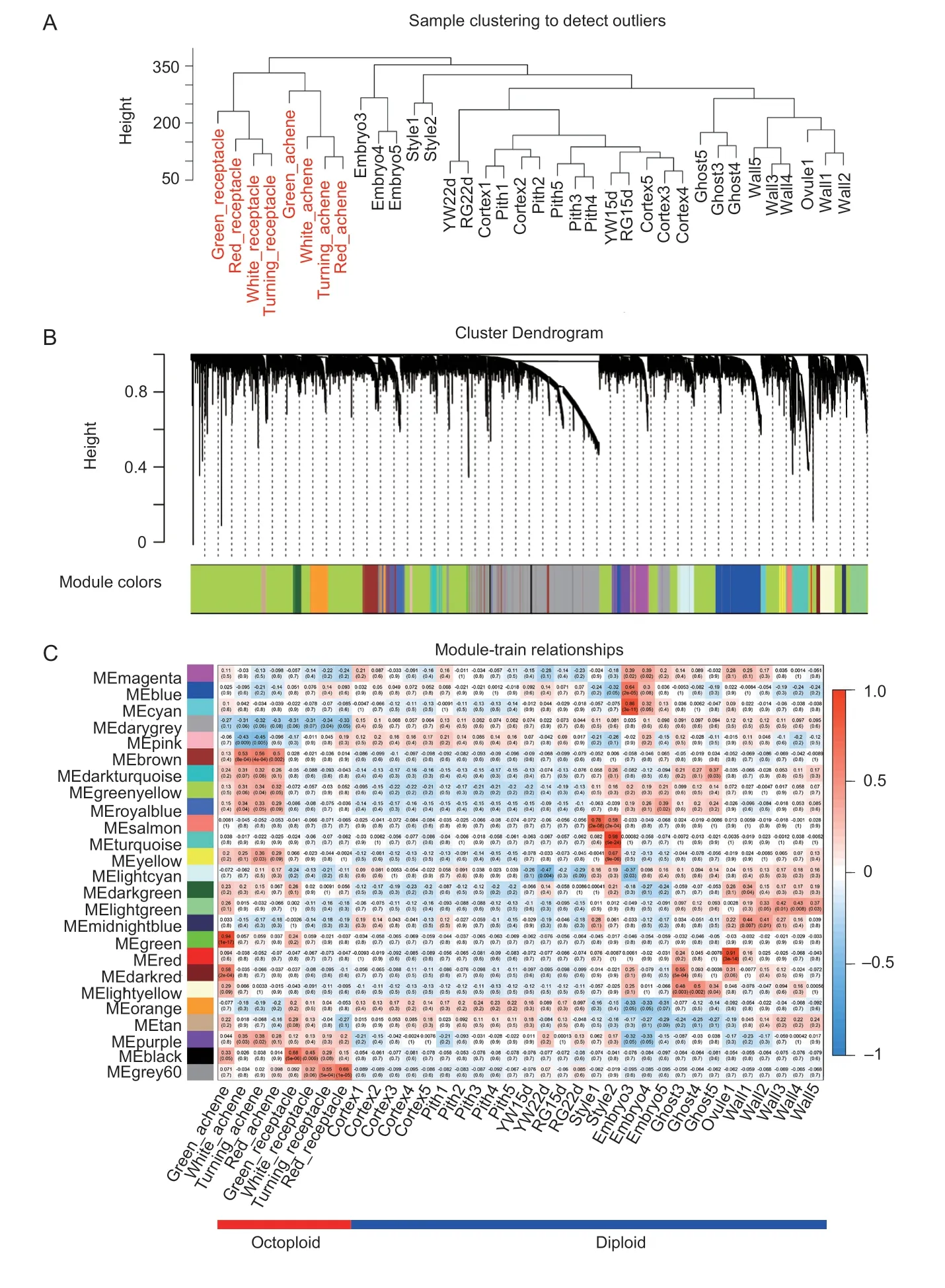
Fig.3 Coexpression network analyses of flower,fruit and vegetative tissues. A,cluster dendrogram showing the global relationship between different samples. B,dendrogram showing co-expression modules identified by WGCNA. Each leaf in the tree is one gene. The major tree branches constitute 26 modules labeled by different colors. C,a heatmap showing cluster-tissue associations of the network. Each row corresponds to a module. Each column corresponds to a specific tissue/stage. The color in each box indicates the correlation coefficient between the module and tissue type.
3.5.Receptacle-associated modules provide insight into metabolic regulation network during fruit ripening
The most obvious phenomenon during fruit ripening is the synthesis of anthocyanins. Previous transcriptome analyses revealed that the transcription of flavonoid biosynthetic genes was induced in the receptacle,implicating the importance of these genes (Hawkinset al.2017). The grey60 color module (Appendix E) is correlated with the development of the octoploid receptacle,and the correlation increases as the receptacle matures (Fig.3-C).Fig.4-A shows the eigengene expression of the grey60 module. A heatmap shows the relative TPM of genes from the receptacle module and revealed that many genes were highly expressed in both the octoploid ripening fruit(Fig.4-B) and diploid (Yw22D and Rg22D). This result indicates that this module is highly associated with fruit ripening in either diploid or octoploid fruit. WGCNA was also utilized to construct gene networks,and the networks were visualized using Cytoscape (Fig.4-C). Two MYB genes(FvH4_3g1399 and FvH4_3g13990) were found among the genes. FvH4_1g22020 (FvMYB10) can regulate flavonoid/phenylpropanoid metabolism during strawberry fruit ripening(Medina-Pucheet al.2013). FvH4_3g13990 is a homolog of theArabidopsisAtMYB67(AT3G12720)gene,which is a member of the R2R3-MYB factor gene family. The gene ontology (GO) terms (Biological Process) in the grey60 color module are“lipid metabolic process,”“transferase activity,”“carbohydrate binding”and“catalytic activity”(Fig.4-D,Appendix F). The KEGG analysis showed that metabolismrelated pathways were significantly enriched among the grey60 genes,including“metabolism,”“metabolism of terpenoids and polyketides”and“pyrimidine metabolism”(Fig.4-E).
3.6.Species-specific modules and hub genes were identified using WGCNA
Of particular interest is the identification of the speciesspecific gene modules owing to the many differences in octoploid and diploid fruits. We used samples from all the stages of fruit development and performed a WGCNA analysis to identify conserved genes across species.Notably,six out of 26 coexpression modules (darkgrey,brown,green,purple,black and grey60;P<0.001) were composed of genes that were highly expressed in a single species (Appendix G). Among them,the darkgrey and black color modules were the most significant module for each species. Fig.5-A and B show the eigengene expression for the darkgrey (diploid) module and the black (octoploid)module,respectively.
The darkgrey (diploid) module (Fig.5-A,Appendix H)exhibits a positive correlation with diploid fruit tissues and a negative correlation with octoploid fruit tissues. We performed the GO enrichment analysis to detect the ability of the consensus clusters to predict functional relationships between genes. The GO terms (Biological Process)in the diploid (darkgrey) module were related to“cell communication,”“signal transduction”and“cellular process”et al.(Appendix F). We also used WGCNA (Langfelder and Horvath 2008) to construct gene networks,in which each node represents a gene,and the connecting lines (edges)between genes represent coexpression correlations.WGCNA identifies the most highly connected,or the most central genes,within each module,referred to as“hub genes.”The top 10 genes in each of the specific modules are shown in Table 1.
In the darkgrey (diploid) module network (Fig.5-C;Appendix H),many of the network genes are F-box genes. Interestingly,a previous study showed that the F-box genes and miRNA formation of the miRNA-FBX networks inF.vescafruit and FBX-targeting miRFBXs could have evolved to regulate processes specific toF.vesca(Xiaet al.2015). In addition,the hub genes in the diploid module include BAK1 (FvH4_1g04660),ataurora3(FvH4_1g06230) and two TIR-NBS-LRR (FvH4_1g07010 and FvH4_1g07020)et al.(Table 1).
In contrast,the black (octoploid) module exhibits a positive correlation with octoploid fruit tissues (Fig.5-B,Appendix I) and a negative correlation with diploid fruit tissues. The GO terms (Biological Process) in the black module are related to“cellular protein modification process”(Appendix F). Strikingly,many wall-associated kinases(WAKs) genes were in the octoploid (darkgrey) module network (Fig.5-D). The top 10 list of hub genes includes one WAK gene,which could be involved in cell wall-related processes (Table 1).
4.Discussion
4.1.A large number of genes exhibit differentially regulated AS events between the diploid F.vesca and octoploid F.×ananassa
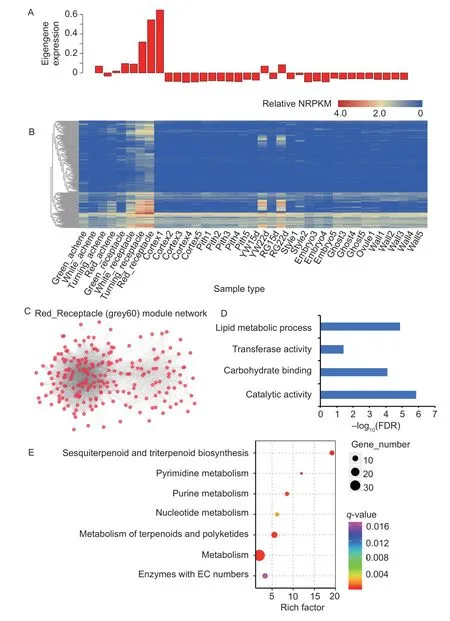
Fig.4 Ripe fruit-associated genes and networks. A,eigengene expression profile for the ripe-fruit module in different tissues.B,the correlation network of the ripe-fruit module. Each colored circle (node) represents one gene. C,the correlation network of the ripe-fruit (grey60) module. D,GO terms are enriched in the ripe-fruit module genes. E,the enrichment analysis of KEGG pathway for the genes in the ripe-fruit module. The area of each colored circle is proportional to the number of genes involved in each pathway,and the color indicates the q-value.
The genusFragariais composed of both cultivated(F.×ananassa) and wild species with various ploidy levels,including diploid,tetraploid,hexaploid,octoploid and decaploid (Listonet al.2014). Thus,it is an ideal genus for ploidy research.Fragaria vescais the progenitor diploid species ofF.×ananassa,and the quality of their fruits differ in terms of size,color,aroma and texture. Therefore,comparing the transcriptome landscapes between theF.vescaandF.×ananassareceptacles could provide insights into the regulation of both ploidy and fruit quality.Interesting alterations of AS between diploid and tetraploid varieties in some genes have been observed inBrassicaand watermelon (Zhouet al.2011;Saminathanet al.2015).However,these studies only examined a limited number of genes that would be unlikely to reflect the change of the entire transcriptome. In this study,the overall features of AS are the same between diploid and octoploid plants. For example,the percentages of each major AS type are similar(Fig.2),indicating that the splicing machinery functions in the same manner. TheF.×ananassaspecies originates from the natural hybridization betweenF.virginianaandF.chiloensis. Thus,theF.×ananassa‘Sweet Charlie’ has overcome the transcriptome shock that appeared in the first few generations of allopolyploidy or autopolyploidy (Hegartyet al.2006;Buggset al.2011). We cannot infer the AS features during the transcriptome shock.
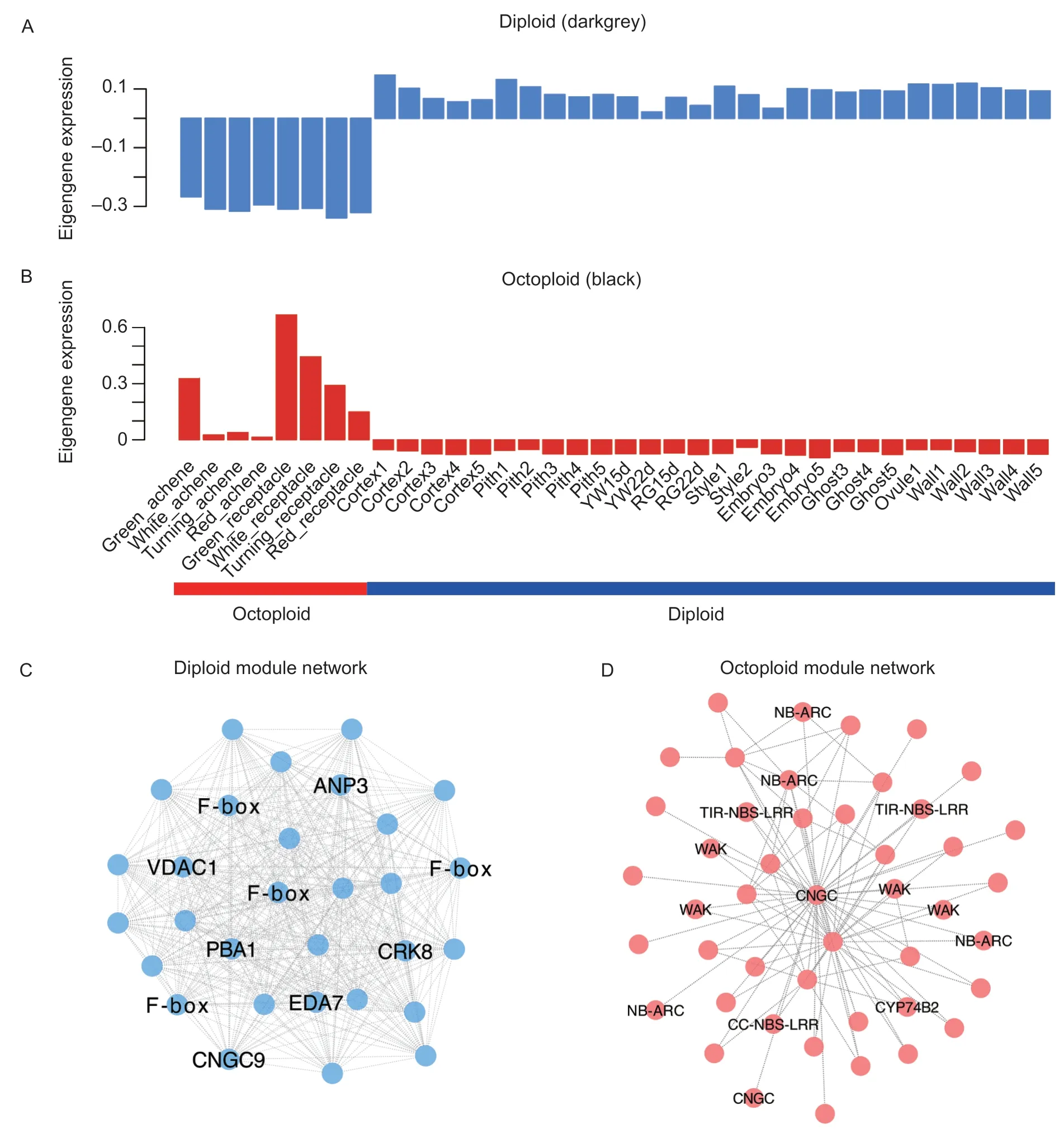
Fig.5 Diploid and octoploid-specific genes and networks. A and B,eigengene expression profiles for the diploid (darkgrey) and octoploid (black) module in different tissues (Correspond to Appendix G). C,the correlation network of the darkgrey (diploid)module. D,the correlation network of the black (octoploid) module. Genes with the edge weight higher than 0.2 are visualized using Cytoscape.
In summary,a large number of genes exhibit differentially regulated AS events between the diploidF.vescaand octoploidF.×ananassa. However,the general AS characteristics are comparable between these two strawberries with different ploidy levels. Altogether,this study identified the majority of alternatively spliced eventsduring receptacle development in strawberry,serving as a valuable resource for future functional studies.
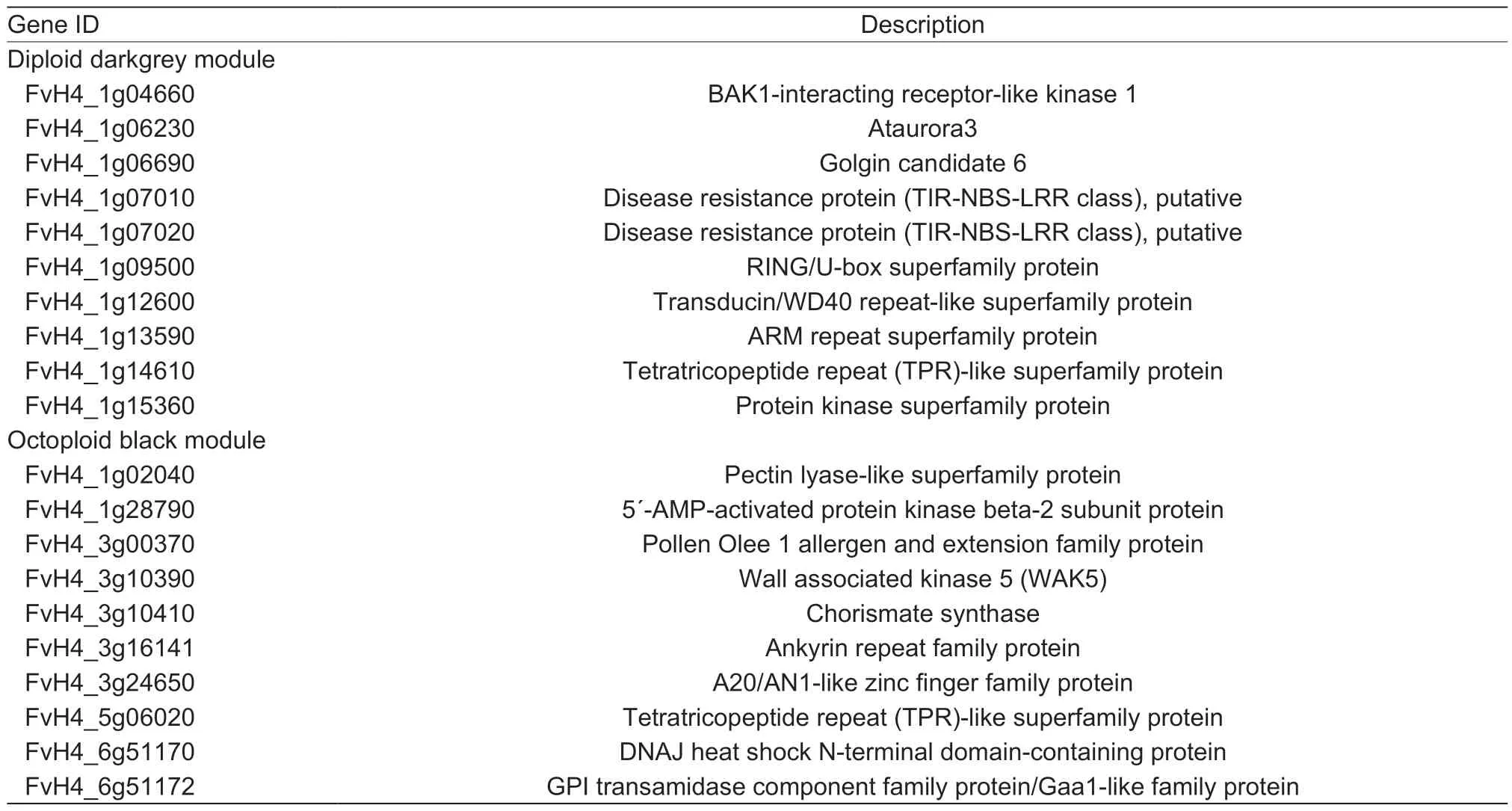
Table 1 Top10 candidate hub genes in diploid and octoploid modules,respectively
4.2.Flavonoid biosynthetic pathway during fruit ripening
Ripening strawberry fruit undergo two flavonoid formation stages. The first stage is the formation of flavanols,and the second is related to the accumulation of anthocyanins and flavonols (Halbwirthet al.2006). Ripe strawberry fruit contain high concentrations of flavonoids,particularly red colored anthocyanins (Almeidaet al.2007). The anthocyanins are primarily present in the form of PAs before the fruit ripen. The levels of PAs will subsequently decrease considerably during fruit ripening (Carboneet al.2009;Schaartet al.2013).
In this study,the standard networks were generated by the WGCNA analysis pipeline. Our network analyses revealed five flavonoid biosynthesis-related genes (FvGST,FvMYB10,FvMYB9/11,FvUFGTandFvH4_1g02120) in the ripe-fruit module (Fig.4),which prompted us to investigate the flavonoid biosynthetic pathway during the ripening of fruit. In strawberry,FaMYB9/FaMYB11,FabHLH3andFaTTG1are homologs ofArabidopsisAtTT2,AtTT8andAtTTG1,which form a ternary protein complex to regulate the expression of the PA biosynthetic genes (Schaartet al.2013). The expression ofMYB10,a R2RMYB transcription factor,is primarily repressed in the fruit receptacle. However,it is activated by ABA and repressed by auxins during the process of fruit ripening. It plays an important regulatory role in the flavonoid/phenylpropanoid pathway during the ripening of strawberry (Medina-Pucheet al.2013). A reduced anthocyanin in petioles (rap) mutant was identified to be a premature stop codon in a GSTFvGSTgene.FvGSTmediates foliage and fruit pigmentation and also acts downstream of the fruit-specificFvMYB10(Luoet al.2018).InArabidopsis,the homolog of strawberryFvUFGTencodes an anthocyanidin 3-O-glucosyltransferase,which is involved in the biosynthesis of flavonoid glucosides. FvH4_1g02120,which is a homolog of theArabidopsisAT1G49390gene,was also involved in the flavonoid biosynthetic process.Overall,the synthesis of anthocyanins in strawberry fruit involves a highly complex regulatory network.
4.3.Wall-associated kinases (WAKs) may function in defense and the development of octoploid strawberry
It is interesting to note that many WAK genes were found in the octoploid module network (Fig.5-D). The wallassociated kinases (WAKs) are receptor-like kinases,which have a cytoplasmic protein kinase domain and are linked to the pectin fraction of the plant cell wall. The plant cell wall is primarily composed of cellulose,hemicellulose,pectin and proteins. The wall is thought to be involved in modulating plant development (Kohorn 2000). Cell expansion(Cosgrove 1997) and pathogen infection (Hammond-Kosack and Jones 1996) can alter biosynthetic and cell wall components and downstream cytoplasmic events such as systemic acquired resistance. These events show that communication between the cytoplasm and cell wall is very important. WAKs are one of the four classes of proteins that physically link the ECM to the plasma membrane and may promote communication between these two compartments.It is well known that cultivated strawberry (F.×ananassa)is an allopolyploid (octoploid) species with fruit that are large in size,disease resistant,and have intense color and varied aromas and flavors compared with diploid varieties. This is consistent with the many WAK genes that exist in the octoploid module network. Since WAKs proteins regulate diverse biological processes,they could function to communicate damage signals and modulate both plant defense and development (Kohornet al.2009;Brutuset al.2010). FBX proteins regulate diverse biological processes and are grouped into two general types based on whether they function in conserved processes,such as embryogenesis and circadian rhythms,or relatively specialized processes,such as pollen recognition and pathogen response. A novel miRNA-FBX network has been identified inF.vesca. The miRFBX could also promote the accumulation of resistance proteins and enhance disease resistance inF.vescafruit (Xiaet al.2015).
5.Conclusion
In this study,a large number of genes were differentially expressed at the different stages of fruit development betweenF.vescaandF.×ananassa. The genes involved in flavonoid biosynthesis were concerned with fruit ripening.The species-specific regulated network existed in the octoploid and diploid fruit,respectively. We found that theWAKandF-boxgenes were enriched in the octoploid and diploid fruits,respectively. This study will be a valuable resource for future flavonoid biosynthetic and fruit size studies in strawberry.
Acknowledgements
This work was supported by the Program for High-level University Construction of the Fujian Agriculture and Forestry University,China (612014028),the Natural Science Foundation of Fujian Province,China (2018J01700) and Rural Revitalization Service Team of Fujian Agriculture and Forestry University,China (11899170125).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- Low glycemic index:The next target for rice production in China?
- Do cooperatives participation and technology adoption improve farmers’ welfare in China? A joint analysis accounting for selection bias
- Impacts of household income on beef at-home consumption:Evidence from urban China
- The water-saving potential of using micro-sprinkling irrigation for winter wheat production on the North China Plain
- Changes in bacterial community and abundance of functional genes in paddy soil with cry1Ab transgenic rice
- Synergistic effect of Si and K in improving the growth,ion distribution and partitioning of Lolium perenne L.under saline-alkali stress