C型凝集素样受体-2在血栓形成中的作用研究进展
2021-05-21徐士雪杨阳何杨兰祁兴顺
徐士雪,杨阳,何杨兰,祁兴顺
1北部战区总医院消化内科,沈阳 110840;2中国医科大学研究生院,沈阳 110013;3西北大学生命科学学院,西安710069
C型凝集素样受体-2(C-type lectin-like receptor 2,CLEC-2)是细胞表面受体C型凝集素超家族的一员[1],其信号转导依赖于胞质的免疫受体酪氨酸活化基序(immunoreceptor tyrosine-based activation motif,ITAM),每个ITAM包含2个保守的YXXL基序,但CLEC-2只拥有1个YXXL基序,故2个磷酸化的受体之间通过脾酪氨酸激酶(spleen tyrosine kinase, syk)中串联的SH2结构域进行桥接形成二聚体,1个syk通过结合2分子磷酸化的YXXL进行酪氨酸磷酸化。syk激活后,其下游含有SH2结构域的76 ku的白细胞蛋白(SH2 domain-containing leukocyte protein of 76 ku,SLP-76)及磷脂酶γ2(PLCγ2)被激活,使钙离子浓度增加,血小板活化[2](图1)。在人体内该过程高度依赖于血小板衍生的二磷酸腺苷(ADP)及血栓烷A2(TXA2)施加的正反馈[3]。CLEC-2在血小板及巨核细胞中高表达,在肝Kupffer细胞中低表达。平足蛋白(podoplanin,PDPN)是其目前已知的唯一内源性配体,但在血管内皮细胞中并不表达[2]。
血栓形成是人体内的重要保护机制,当血管或组织损伤时,血栓形成可以避免机体过度失血,但同时它也是缺血性心脏病、缺血性脑卒中及静脉血栓栓塞等血栓栓塞性疾病的病理基础。早期研究发现,CLEC-2特异性缺失的小鼠均有血栓形成障碍表现,进而发现CLEC-2可介导血小板活化及聚集[4];另一方面,表达于巨核细胞的CLEC-2介导了血小板生成素的产生,并可通过促进PDPN分泌趋化因子5(CCL5)而导致巨核细胞前血小板化[5]。以上研究结果表明,CLEC-2表达与血栓稳定及聚集有密切关系。本文从动脉粥样硬化、炎症血栓状态、血管壁完整性、癌症相关血栓等4个方面对CLEC-2在血栓形成过程中的作用进行综述。

图1 CLEC-2介导血小板活化的示意图Fig.1 Schematic diagram of CLEC-2 mediated platelet activation
1 CLEC-2与动脉粥样硬化
在血流剪切力作用下,动脉粥样硬化斑块破裂导致的血小板聚集是动脉血栓的最常见原因。晚期动脉粥样硬化患者的斑块破裂后,表达在斑块内部的PDPN得以暴露,与血小板表面的CLEC-2结合,促进了血小板活化,进而加速了动脉血栓的形成[6]。Inoue等[7]研究发现,在斑块未破裂时,仍有配体与CLEC-2相互作用促进血栓形成,并通过蛋白芯片等技术证实S100钙结合蛋白A13(S100A13)在粥样硬化早期或某些病理状况下表达于平滑肌细胞的管腔面,通过CLEC-2激活血小板,从而促进血栓形成;此外,行使CLEC-2细胞外域功能的重组CLEC-2还可与正常动脉壁的平滑肌细胞结合,而正常动脉壁并不表达PDPN,也不表达S100A13,因此正常血管壁上仍可能存在其他尚未被发现的CLEC-2配体。Furukoji等[8]发现,血管内皮生长因子-A(vascular endothelial growth factor-A,VEGF-A)可诱导PDPN在动脉血管内皮细胞内表达。动脉粥样硬化发生时,平滑肌细胞及巨噬细胞均可表达VEGF-A,诱导PDPN的过度表达并激活CLEC-2,导致血栓形成。这为CLEC-2在动脉粥样硬化中介导血栓形成的作用机制提供了一种新解释。当动脉粥样硬化斑块破裂时,胶原蛋白也得以暴露,作为最有效的血小板活化物质,胶原蛋白对血小板活化的影响较CLEC-2更强烈[9]。以上结果提示CLEC-2在动脉粥样硬化晚期血栓形成中的作用可能有限。
近年来,越来越多的研究证实了CLEC-2在心脑血管疾病中的临床意义。血浆CLEC-2水平增高与冠状动脉疾病[10]及急性缺血性脑卒中[11]发生风险独立相关。血浆CLEC-2水平增高与卒中加重[优势比(OR)=1.97,95%置信区间(CI)=1.11~3.50,P=0.021]及预后不良(OR=1.70,95%CI=1.17~2.48,P=0.006)显著相关[11]。一项纳入352例急性缺血性脑卒中患者的前瞻性研究发现,在一年随访期间,经对数转换后的血浆CLEC-2水平每增加一个标准差,患者死亡风险增加4.27倍,死亡/心脑血管事件的合并风险增加2.42倍[12]。进一步研究发现,PDPN与CLEC-2在缺血/再灌注损伤的神经元及小胶质细胞中的表达水平均显著上调,而PDPN抗体预处理可明显减轻神经功能损伤并减少梗死面积,提示CLEC-2与PDPN通过参与缺血性脑卒中诱导脑损伤,进而导致预后不良[13]。
2 CLEC-2与炎症血栓状态
血流淤滞及缺氧是静脉血栓形成的始动因素,而血流畸变可导致缺氧,刺激血管内皮细胞内的Weibel-Palade小体释放血管性血友病因子(von Willebrand factor,vWF),导致血小板及中性粒细胞黏附,从而引起血栓及炎症[14]。Payne等[15]建立了模拟静脉瓣膜内血流畸变的下腔静脉模型,48h后,诱导CLEC-2缺失的小鼠均未发生深静脉血栓(deep venous thrombosis,DVT),而未诱导CLEC-2缺失小鼠的DVT发生率高达60%;CLEC-2基因敲除小鼠的DVT发生率也显著低于CLEC-2基因未敲除的健康小鼠(38% vs. 81%),向血小板基因缺陷小鼠输注野生型小鼠的血小板后,血栓形成有所减少,充分证实了CLEC-2(尤其是血小板CLEC-2)在DVT形成中的重要作用;CLEC-2缺失小鼠的血管壁血小板聚集水平较野生型小鼠低(1.3%±0.4% vs. 7.8%±0.7%,P<0.003),而PDPN特异性缺失小鼠的血管壁血小板聚集水平也较野生型小鼠低(2.6%±0.4% vs.6.5%±1.3%,P<0.03),表明CLEC-2及PDPN表达可能是静脉血栓中血小板募集的必要条件。但在生理状态下,PDPN并不在静脉壁表达。
在DVT形成过程中,缺氧导致血压升高及血管壁拉伸,使得内皮细胞间的连接存在缝隙,从而允许血小板渗透到内皮下的空间,因此血小板表面的CLEC-2有机会与PDPN相互作用[15]。有研究在伤寒沙门菌感染小鼠肝脏模型中也发现了类似的促血栓形成机制:在感染后的小鼠肝脏中,多种巨噬细胞的PDPN表达上调,且血管内皮连续性中断的区域可见血小板向肝实质内形成小突起,其机制可能是内皮损伤暴露了表达在肝实质内巨噬细胞中的PDPN,从而促进了CLEC-2依赖的血小板活化[16]。炎症状态导致PDPN上调的机制仍未明确,需进一步研究。
3 CLEC-2与血管壁完整性
血管壁完整性受损会导致内皮下细胞外基质暴露,引起血小板活化并聚集,这一过程可能导致血栓形成[9]。因此,维持血管壁完整性也是避免血栓形成的一个方法。血小板维持血管壁完整性主要是通过ITAM通路进行的,参与这一过程的受体只有糖蛋白(GP)Ⅵ及CLEC-2[17-19]。Boulaftali等[20]利用转基因技术建立了ITAM信号通路选择性缺失的小鼠模型,发现GPⅥ或CLEC-2缺失均在一定程度上减轻了免疫复合物诱导的皮肤炎症,以及脂多糖诱导的肺部炎症中血小板维持血管完整性的能力;GPⅥ与CLEC-2同时缺失的小鼠及ITAM基序下游衔接蛋白SLP-76缺失的小鼠均表现为血小板维持血管完整性功能的完全丧失。作为CLEC-2唯一的内源性配体,PDPN并不表达于血管内皮细胞或周细胞。因此,在脂多糖诱导的肺部炎症中,血小板CLEC-2可能与肺泡Ⅰ型上皮细胞中高表达的PDPN相互作用;在免疫复合物诱导的皮肤炎症中,PDPN可能通过浸润巨噬细胞“传递”到炎症部位,与血小板CLEC-2相互作用,也可能这些组织或血管壁中存在尚未被发现的CLEC-2配体[20]。表达于高内皮微静脉(high endothelial venule,HEV)周围成纤维网状细胞的PDPN与血小板CLEC-2相互作用对维持HEV的完整性至关重要[21]。HEV是淋巴组织中特殊的毛细血管后微静脉,是淋巴细胞自血液进入淋巴组织的通道。虽然PDPN在淋巴管内皮细胞中表达,但并不在包括HEV在内的血管内皮中表达,缺乏CLEC-2或PDPN的小鼠均表现为自发性血管渗血[22]。以上研究证实了血小板CLEC-2的表达对维持血管完整性的重要作用,其机制可能与PDPN有关,也可能与尚未被发现的CLEC-2内源性配体有关。
4 CLEC-2与癌症相关血栓
血小板可促进肿瘤生长或释放生长因子促进肿瘤血管生成,恶性肿瘤也可导致血小板活化,致使机体呈高凝状态[23]。生理状态下,由于剪切应力及免疫细胞的活动,大多数肿瘤细胞不能在血流中存活。然而,由于多种肿瘤细胞表面表达PDPN,血小板通过CLEC-2与PDPN结合包裹在肿瘤细胞表面,不仅可帮助肿瘤细胞逃避机体的免疫监视,还可使肿瘤细胞免受血流剪切应力的影响[24],使循环中的肿瘤细胞增多,导致癌性血栓发生率增高[24-25]。Shirai等[23]研究发现,CLEC-2缺失小鼠肿瘤血管中的血栓形成减少,肺栓塞发生率降低。一项纳入了285例恶性脑肿瘤患者的前瞻性研究证实,肿瘤细胞PDPN阳性表达患者的血小板计数降低、D-二聚体水平升高,PDPN高表达患者的半年、1年、2年累积静脉血栓发生率分别为18.4%、25.7%、25.7%,均高于PDPN低表达患者(分别为8.7%、10.3%、14.6%)[26]。以上研究证实CLEC-2与PDPN相互作用能明显影响癌症患者的预后。目前已有研究尝试通过研发干预CLEC-2与PDPN相互作用的药物来抑制肿瘤转移及癌性血栓形成,进而改善患者的生存状况[27]。
5 总结与展望
CLEC-2可介导多种病理状态下的血栓形成(图2),但CLEC-2特异性缺失并不影响正常止血[28],这将为许多血栓栓塞性疾病提供新的治疗手段。目前研究仍存在不足:(1)目前的研究多为动物模型研究,尚欠缺临床研究。(2)小鼠与人体的CLEC-2表达有所差异。人体中CLEC-2只存在于血小板、巨核细胞及肝脏中;小鼠中CLEC-2还表达于一些免疫细胞,包括树突细胞、外周中性粒细胞、炎性巨噬细胞、自然杀伤细胞及B细胞[2]。(3)小鼠血小板表达ITAM通路的受体只有GPⅥ及CLEC-2,而人类血小板还包含蛋白质人免疫球蛋白G Fc段Ⅱ型受体(FcγRⅡA)[15],故小鼠实验所得结论未必适用于人体。未来可从以上三个方面或通过其他作用机制开展进一步研究,以使CLEC-2更有效地应用于临床实践,发挥更好的治疗作用。
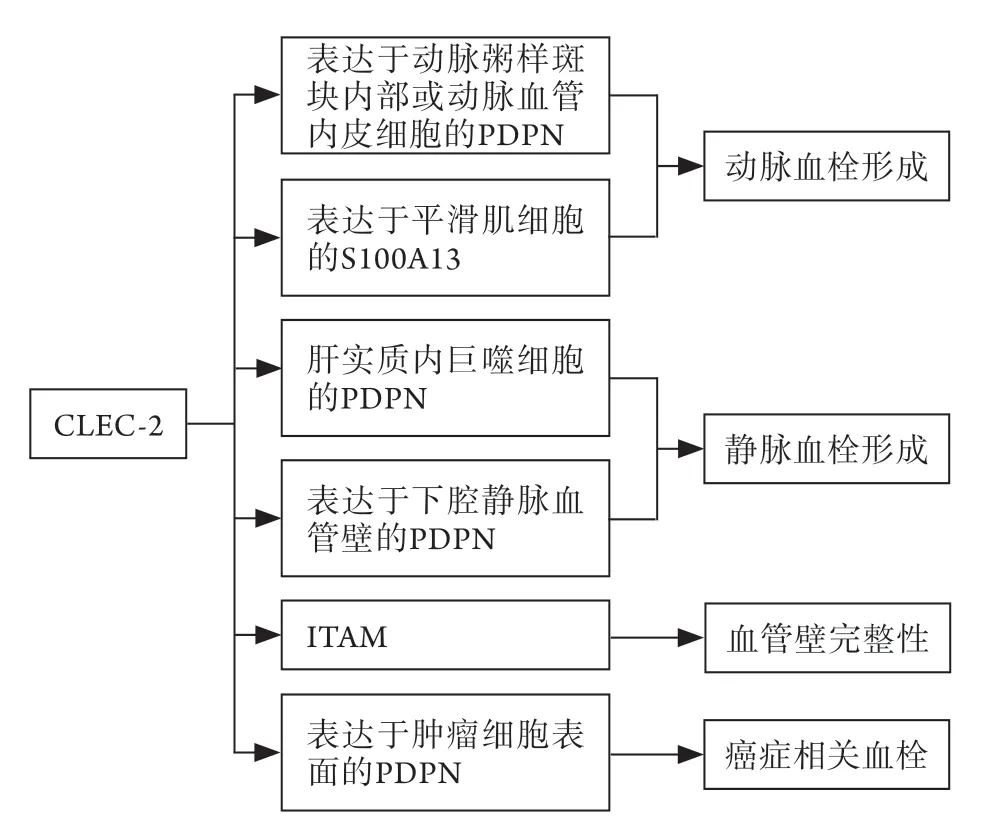
图2 CLEC-2介导血栓形成的主要途径Fig.2 Major pathways of CLEC-2 mediating thrombosis
