基于DFT 计算的1,4-丁二醇脱水反应过程分析
2021-05-19米容立吴志强杨伯伦
米容立, 吴志强, 杨伯伦
(1. 西安交通大学 化学工程与技术学院, 陕西 西安 710049;2. 西安交通大学 动力工程多相流国家重点实验室, 陕西 西安 710049)
1 前 言
1,4-丁二醇(1,4-butanediol,BDO)是一种含有4 个碳原子的二元伯醇,主要用于生产包括四氢呋喃(tetrahydrofuran,THF)[1]、γ-丁内酯(gamma-butyrolactone,GBL)[2]、聚对苯二甲酸丁二醇酯(polybutylene terephthalate,PBT)[3]、聚氨基甲酸酯(polyurethane,PU)[4]等在内的一系列精细化学品,是十分重要的化工原料。然而,随着近年来BDO 产业的迅速扩张,整个行业步入供过于求的时代[5]。探索更多的BDO下游产品生产工艺,扩展BDO 产业链,缓解产能过剩所带来的风险是目前急需解决的问题。3-丁烯-1-醇(3-butene-1-ol,BTO)是一种高价值的不饱和醇类化合物,常用作医药、农化产品以及食品添加剂等产品的中间体,尤其在新型杂环类药物、抗肿瘤药物、抗艾滋药物的合成中具有不可替代的地位[6-7]。从BDO 脱水制备BTO 不但为BTO 规模化生产提供了新工艺,同时对BDO 下游产业链的扩展也具有重要的意义。然而,BDO 脱水是一个较为复杂的反应网络,能够发生包括脱水、脱氢、异构化和加氢等多种反应[8],各反应或相互竞争,或相互促进,共同决定了BDO 脱水反应的产物分布。然而,目前的BDO脱水反应研究主要集中于催化剂的开发,对于反应机理的研究却很少[9-12]。
反应体系的热力学分析、微观反应动力学研究对于催化剂的设计、反应器的选型、工艺流程的优化乃至反应过程的强化具有十分重要的意义。王丽苹等[13]采用Benson 和Joback 基团贡献法对碳酸二甲酯与BDO 合成聚碳酸酯二醇的反应体系进行了热力学分析,计算了BDO 在不同温度下的生成焓、摩尔等压热容以及标准熵。Qian 等[14]研究了BDO 与环己酮的缩酮反应,并对体系的热力学性质进行了分析。Ermelinda 等[15]通过实验的方法报道了BDO 不同异构体之间的蒸发焓。Zorebski 等[16]测量了不同温度下BDO 的摩尔热容。这些研究虽然涉及了BDO 部分热力学性质的计算和测量,但本反应体系中所涉及的3-丁烯-1-醇、反-2-丁烯-1-醇、顺-2-丁烯-1-醇、γ-丁内酯等物质的热力学数据却未见报道,无法直接进行BDO 脱水反应网络热力学分析,可采用量子化学的方法对反应的热力学及其微观机理进行理论研究。Alexopoulos 等[17]采用密度泛函理论研究了乙醇脱水反应网络,不仅分析了乙醇脱水生成乙烯和乙醚的反应路径,还提出了乙醚进一步分解生成乙烯和乙醇的反应机理,并计算了相应的动力学数据。John 等[18]采用第一性原理对丁醇脱水反应进行了研究,提出了反应网络中各反应的反应机理,并通过过渡态理论对相应的动力学参数进行了计算。同时,John 等还发现,提高反应温度并降低丁醇分压有利于其脱水生成丁烯。反之,则有利于分子间脱水产物二丁醚的生成。
基此,本文将通过量子化学的方法,对反应体系中各反应物和产物进行结构优化和频率计算,得到BDO 脱水反应网络中各反应在不同温度下的热力学参数,并以此对反应体系进行热力学分析。同时,对各反应的过渡态及反应路径进行搜索,确定反应的微观机理。以变分过渡态理论为基础,对反应体系中各反应的动力学性质进行研究,以期为该反应体系催化剂的设计以及工艺参数的优化提供理论依据。
2 计算方法
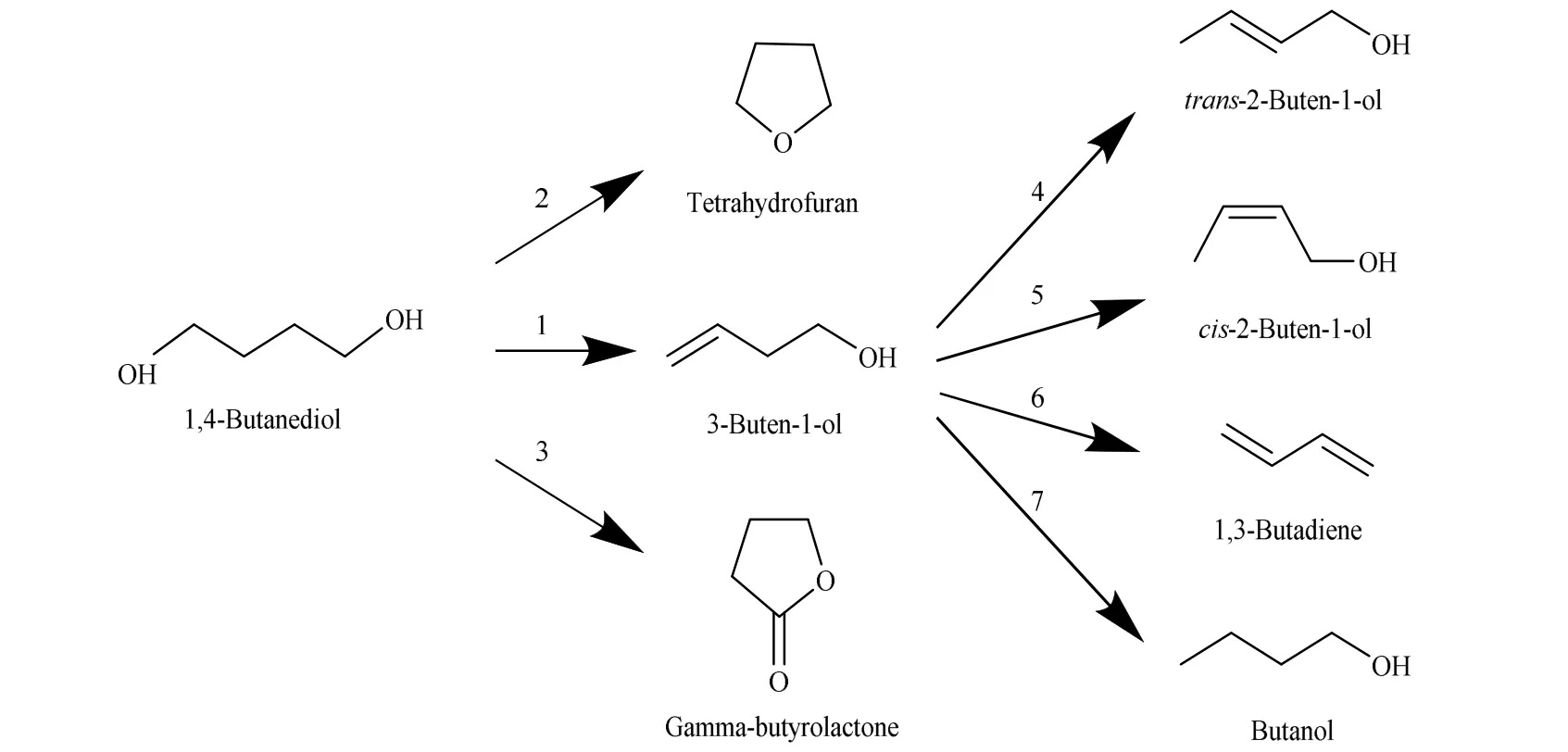
图1 BDO 脱水反应网络Fig.1 Reaction network of BDO dehydration
BD O 脱水反应网络如图1 所示,主要包括7 个反应,即BDO 脱水生成BTO 的主反应(反应1),BDO环化脱水生成THF(反应2)和BDO 脱氢生成GBL(反应3)的2 个平行副反应,BTO 发生双键异构化生成反-2-丁烯-1-醇(trans-2-buten-1-ol, T-2BT1O,反应 4)和同分异构体顺-2-丁烯-1-醇(cis-2-buten-1-ol,C-2BT1O,反应5),以及BTO 脱水生成1,3-丁二烯(1,3-butadiene, BDE,反应6)和BTO 加氢生成丁醇(butanol,BuOH,反应7) 4 个串联副反应[3,5-6,8]。
本研究利用密度泛函理论通过Gaussian 09 软件包对上述反应进行量子化学计算[19-20]。采用m06-2x泛函,6-311g+(d, p)基组对反应体系中各物质进行几何结构优化和频率分析,以得到各物质的零点能、焓和熵。同时采用更高级别的def2-TZVPP 基组对优化后的构型进行单点能计算,得到优化构型的电子能量E。这一组合方法具有较高的性价比既能保证计算精度又能节约计算成本[21-22]。各物质的Gibbs 自由能可由式(1)计算得到

式中:ΔH 为该物质目标温度及绝对零度下焓值的差值(H(T)-H(0))。同时,采用Alecu 等[23]拟合的谐振频率校正因子对各物质的热力学性质进行校正。并通过式(2)~(4)计算各反应的热力学参数:
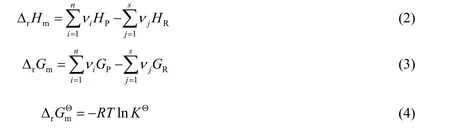
通过对反应物和产物的几何结构进行分析,猜测初始的过渡态结构并通过TS(Transition State)方法对各反应可能的过渡态进行搜索。对所得过渡态的结构进行频率计算,验证其虚频的个数,并进行自然键轨道 (natural bond orbital,NBO)分析。随后,通过内禀反应坐标(intrinsic reaction coordinate,IRC)的计算获取反应过程中的最小能量路径(minimum energy path,MEP),并以此来判断过渡态结构的合理性[24]。
基于反应路径的计算,利用KiSThelP 开源软件(Kinetic and Statistical Thermodynamical Package)[25],以变分过渡态理论为基础计算各反应的反应速率常数:
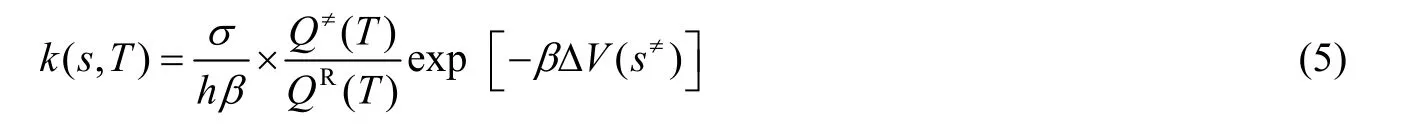
此外,通过Wigner 方法计算隧道校正因子κ,并对反应速率常数进行隧道效应校正。其中Wigner 方法计算如式(6):

考虑隧道效应后的反应速率常数表达式如下:

3 结果与讨论
3.1 1,4-丁二醇脱水反应网络中各反应热力学性质的计算和分析
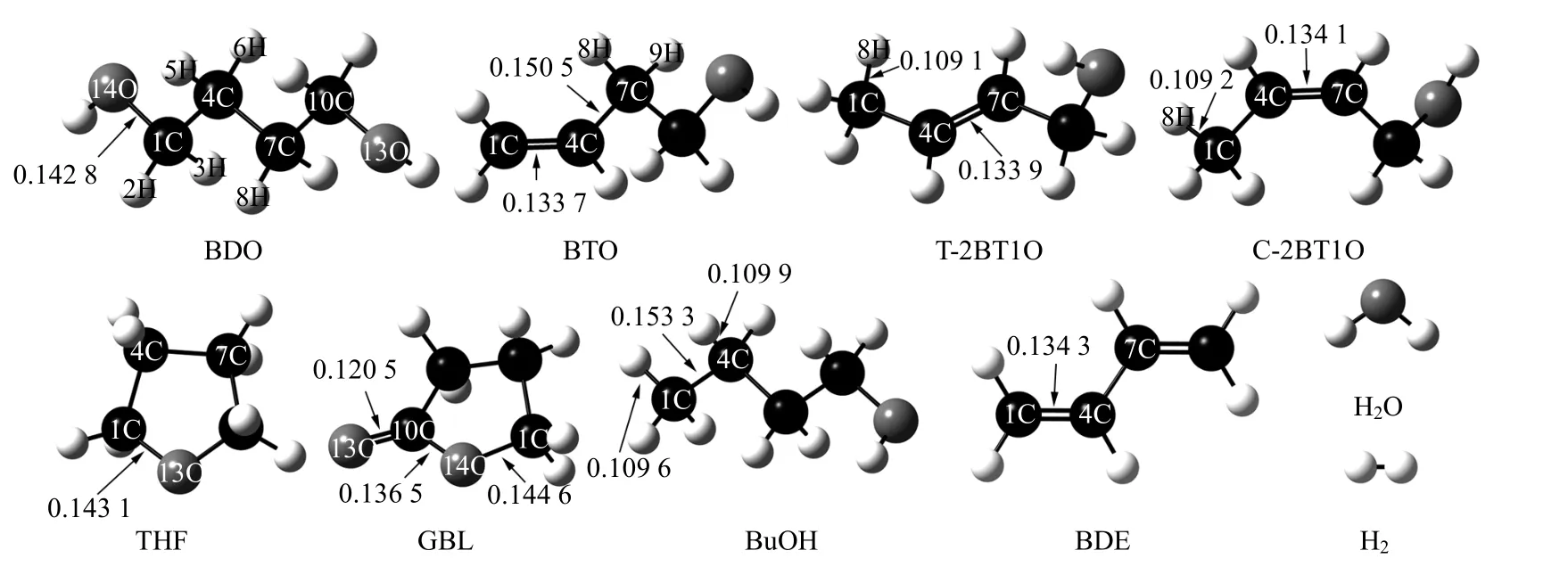
图2 BDO 反应网络中各物质的优化几何结构(键长单位为nm)Fig.2 Optimized geometries of chemicals in BDO reaction network
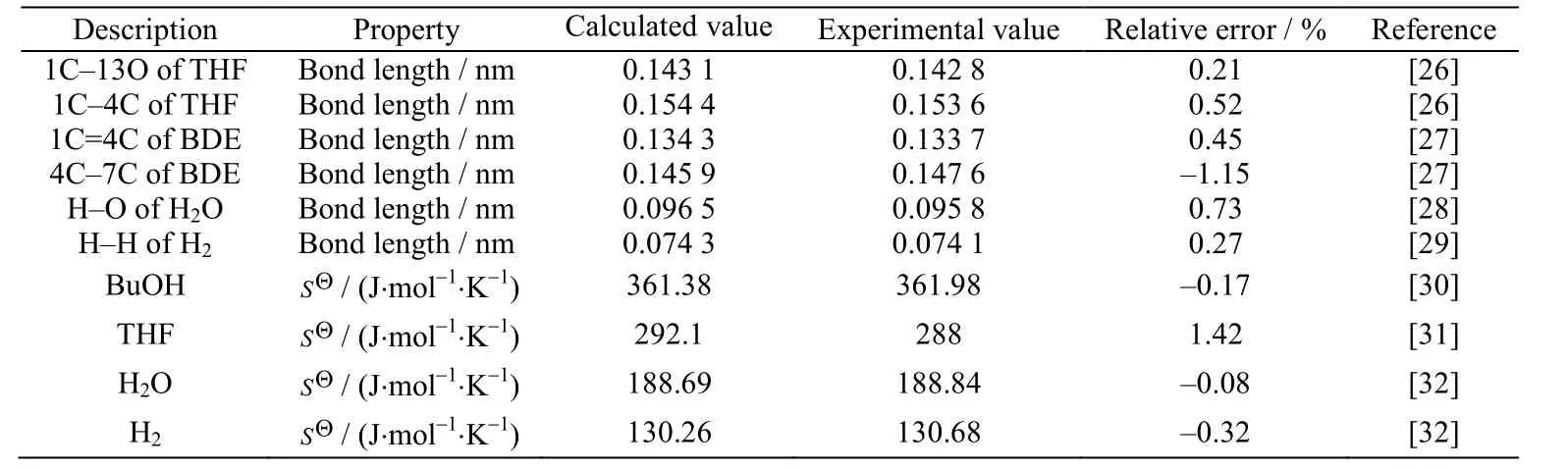
表1 计算结果验证Table 1 Verification of calculated results
本文在m06-2x/6-311g+(d,p)水平上对反应物和产物进行结构优化,优化后各物质的构型如图2 所示,其吉布斯自由能可以由式(1)计算得到。采用Alecu 等[23]报道的谐振频率校正因子对反应物及产物进行校正,并通过式(2)~(4)计算不同反应温度下各反应的热力学数据。其中,部分物质热力学参数、结构的计算值与文献报道的实验值如表1 所示,从表中可见其相对误差最大不超过2%,表明本文的计算方法较为可靠,可以用来对BDO 脱水反应体系进行理论计算。不同温度下各反应的摩尔反应焓如图3 所示。从图中可以看出反应1、反应6 以及反应3 的摩尔反应焓都大于0,均属于吸热反应,但是各摩尔反应焓随温度的变化趋势和程度有所不同。反应3 的吸热量最大,且随着反应温度的升高,摩尔反应焓迅速增加。反应1、反应6 也是吸热反应,但是其吸热量低于反应3,且随着反应温度的升高,摩尔反应焓的变化并不明显。反应2、反应4、反应5 以及反应7的摩尔反应焓都小于0,属于放热反应。其中,在T = 298 K 时,反应7 的反应放热量最大,反应2 放热量最小;随着反应温度的增加,反应2 摩尔焓变的绝对值迅速增加,在T = 800 K 时成为放热量最大的反应。反应4 和反应5 也均为放热反应,其摩尔反应焓的绝对值也随着反应温度的升高而增加。总之,反应1、反应3 和反应6 为吸热反应,反应2、4、5 和7 为放热反应。
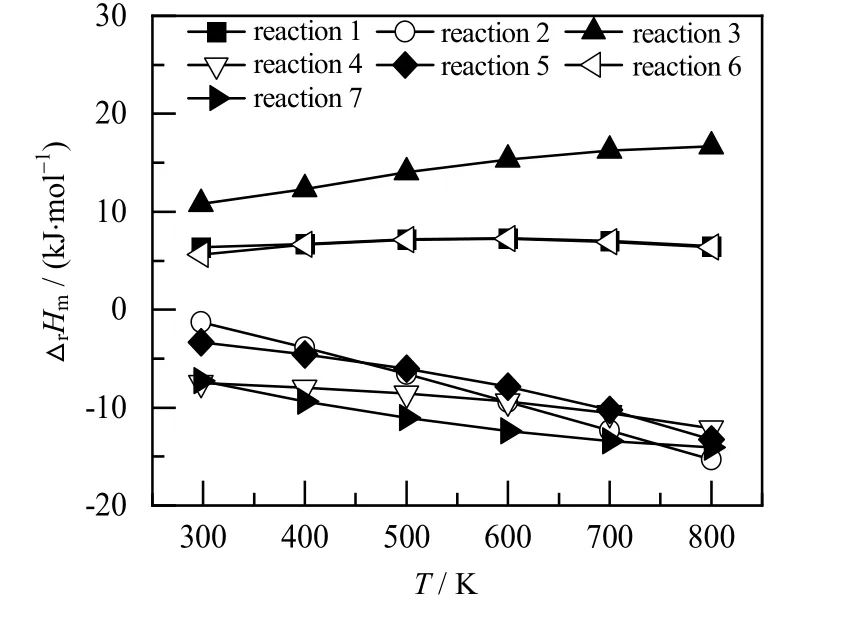
图3 不同温度下各反应的摩尔反应焓Fig.3 Molar enthalpy of reactions at different temperatures
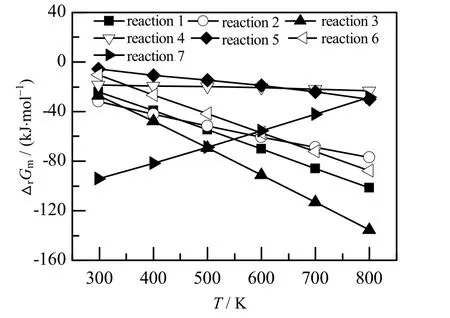
图4 不同温度下各反应的摩尔反应Gibbs 自由能Fig.4 Molar Gibbs free energy of reactions at different temperatures
不同温度下各反应的摩尔反应Gibbs 自由能变如图4 所示。在T = 298~800 K 时,所有反应的摩尔Gibbs 自由能变均小于零,说明其在热力学上都是能够自发进行的[33]。
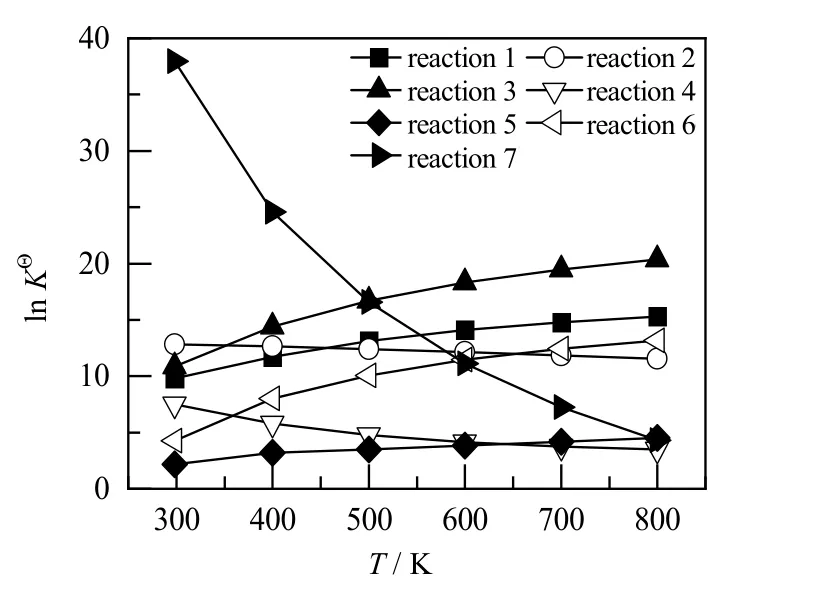
图5 不同温度下各反应的反应平衡常数Fig.5 Reaction equilibrium constants of reactions at different temperatures
BDO 脱水反应网络中各反应平衡常数KΘ与反应温度的关系如图5 所示。在T = 298~800 K 时,反应1、3、6 的反应平衡常数较大,且随着温度的升高,平衡常数进一步增加。因此,温度升高有利于其正反应的进行[34]。反应2、4、7 的平衡常数随温度的升高不断下降,说明高温不利于其正反应的进行。值得注意的是,反应5 虽然是放热反应,但由于有摩尔反应熵及反应温度的影响,其平衡常数随着温度的升高而略有增加,表明高温对其反应的正向进行有利。此外,从图4 和5 中可以看出,反应2 的摩尔反应Gibbs自由能随着反应温度的增加逐渐减小,其平衡常数也是逐渐减小的。这是因为反应平衡常数是通过式(4)计算得到的,当摩尔反应Gibbs 自由能的绝对值增加速率小于反应温度T 的增加速率时,反应平衡常数便呈现出逐渐减小的趋势。
对于BDO 脱水反应网络来说,反应1、2、3 的平衡常数都在较高的水平上,三者都可以达到很高的反应程度。同时,由于反应6 和7 的平衡常数也非常大,在反应达到平衡时,通过反应1 生成的BTO 会继续脱水转化为BDE 等串联副反应产物,从而导致平衡产物中BTO 的含量降低。在低温下,由于生成BuOH 的反应平衡常数远大于其他反应的平衡常数,反应体系中会生成大量的BuOH。随着反应温度的提高,BTO 脱水生成BDE 的反应平衡常数迅速增大,平衡产物中的BTO 进一步反应生成大量的BDE。可以看出,由于各反应的平衡常数都较高,反应程度较大,热力学平衡有利于完全脱水产物BDE 的生成。
尽管各反应的平衡常数随着温度的升高有不同的变化趋势,但是基本上所有反应的平衡常数都非常大,当反应温度大于500 K 时,除BTO 异构化反应之外,所有反应的对数ln KΘ都大于9.2。一般认为,当平衡常数K 大于105(ln KΘ> 9.2)时,反应即认为是不可逆反应[35]。对于复杂反应网络来说,若产物的分布是根据热力学平衡得到,则可称该反应为热力学控制(thermodynamic control);若产物的分布取决于反应速率,则可称为动力学控制(kinetic control),而不可逆反应的主要产物通常是动力学控制的产物[36]。因此,对于BDO 脱水反应网络来说,在研究的温度范围,产物分布主要受动力学控制。要想提高产物中BTO 含量,必须对各反应速率进行控制。因此,对反应网络中各反应路径及其动力学特性的研究是十分必要的。
3.2 1,4-丁二醇脱水反应体系分析
上述分析发现,BDO 脱水反应体系为动力学控制,必须通过提高BTO 生成速率,同时降低平行副反应及串联副反应的速率来实现脱水产物中BTO 含量的提高。为此,有必要对各反应机理及动力学进行分析,以此为催化剂的设计、反应条件的优化等研究内容提供理论支撑。
在优化反应物和产物构型基础上,对各反应进行过渡态搜索和IRC 计算,可以得到各反应过渡态及中间产物的优化结构,如图6 所示。反应过程中的能量变化如图7、8 所示。
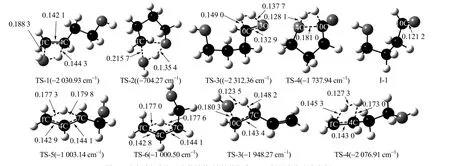
图6 反应过程中各驻点的结构(键长单位为nm;括号内为驻点虚频)Fig.6 Optimized geometries of stationary points in the reaction processes(the bond length unit is nm; imaginary frequency of stationary points is in the brackets)
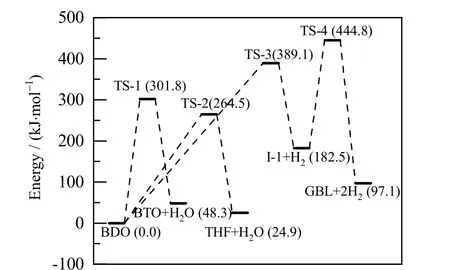
图7 反应1、2、3 的势能剖面图Fig.7 Relative potential energy profiles of reaction 1, 2 and 3 (the potential energy of BDO equals to 0)
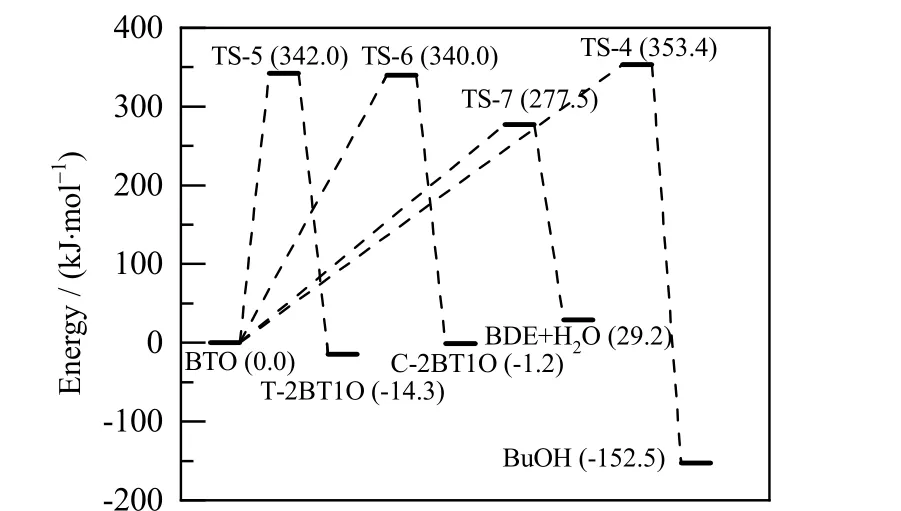
图8 反应4、5、6、7 的势能剖面图Fig.8 Relative potential energy profiles of reaction 4, 5, 6 and 7 (the potential energy of BTO equals to 0)
3.2.1 1,4-丁二醇脱水生成3-丁烯-1-醇
结合图2 及6 可以发现,反应1 是BDO 经由过渡态TS-1 生成BTO 和H2O 的一步反应,该过程需要克服301.8 kJ·mol-1的势垒。反应开始时,5H 向14O 方向移动,5H-4C 和1C-14O 键长因此逐渐伸长,而1C-4C 键长逐渐变短,形成了一个含有1C-4C-5H-14O 四元环的过渡态结构。在此过程中,5H-4C 键长从0.110 1 nm 逐渐伸长到0.144 3 nm;同时,1C-14O 键从0.142 9 伸长到0.188 3 nm,而1C-4C 键长则从0.153 0 减小到0.142 1 nm,并逐渐从单键向双键过渡。14O-5H 距离则缩短到0.126 0 nm,有进一步缩短形成新化学键的趋势。5H 原子上的电子向邻近C 原子及O 原子上转移,本身正电荷增加0.209 6。1C 上的负电荷增加了0.069 7,4C 上的负电荷增加了0.095 3,电子密度的增加使得C-C 逐渐过渡为C=C。同时,5H 上的电子向14O 转移,使得14O 负电荷增加了0.118 2,进而有利于14O 和5H 的化学键的形成。随着反应越过过渡态TS-1,1C-14O 及5H-4C 键长继续伸长,单键逐渐减弱并断裂。同时,1C=4C双键逐渐生成,5H 也继续向14O 方向移动,并最终成键,生成产物BTO 及H2O。
3.2.2 1,4-丁二醇脱水生成四氢呋喃
对反应2 而言,BDO 克服了264.5 kJ·mol-1的势垒形成过渡态TS-2,并进一步生成环化产物THF 和H2O。首先,BDO 分子需要进行构型扭转使得碳链两端的1C 和10C 相互接近。随后,15H 也逐渐向14O移动,二者距离则缩短到0.108 7 nm,1C-14O 因此逐渐伸长到0.215 7 nm。13O 则逐渐向1C 靠近,距离缩短到0.225 8 nm。同时13O-15H 逐渐伸长到0.135 4 nm,形成了一个1C-14O-15H-13O 的四元环过渡态。C-O 键的逐渐断裂以及13O 的靠近导致14O 及1C 上负电荷分别从-0.745 9 和-0.088 0 增加到-0.893 7 和-0.118 5。在14O 和13O 的共同作用下,15H 上的电荷从0.464 3 增加到0.516 1。随着反应越过过渡态TS-2,1C-14O 以及13O-15H 键断裂,新的1C-13O 及14O-15H 键形成,生成了产物THF 和H2O。新的1C-13O 键长为0.143 1 nm,14O-15H 键长为0.096 5 nm。
3.2.3 1,4-丁二醇脱氢生成γ-丁内酯
从图6 和7 中可以看出,反应3 是一个典型的2 步反应,可分别标记为反应3-1 和反应3-2。在反应3-1 中,BDO 首先克服了389.1 kJ·mol-1的势垒形成过渡态TS-3,并脱氢生成中间体I-1;在反应3-2 中,I-1 则通过过渡态TS-4 继续脱氢生成产物GBL,过程中克服了262.3 kJ·mol-1的势垒。
反应过程中,11H 首先向15H 方向移动,键长缩短为0.101 4 nm;10C-11H 及13O-15H 键也随之逐渐伸长,10C-11H 键长从原来的0.110 1 伸长到0.149 0 nm,13O-15H 从原来的0.096 5 伸长到0.137 7 nm;10C-13O 从原来的0.143 0 缩短到0.132 9 nm,其C-O 逐渐由单键向双键转变,进而形成了过渡态TS-3。当反应越过过渡态TS-3 后,10C-11H 及13O-15H 键逐渐断裂,10C-13O 由单键转化为双键,形成中间体I-1。同时,11H-15H 也形成单键,实现了H2的脱除。
随后,中间体I-1 中14O 逐渐靠近10C,二者距离在过渡态时缩短到0.180 8 nm;10C 及14O 上的电子转移至12H 与16H 上,12H 及16H 上的电荷因此分别从0.146 3 和0.478 5 降低到-0.097 4 和0.312 2。同时,10C-12H 以及14O-16H 分别从原来的0.111 0 及0.096 9 nm 伸长到0.160 2 及0.128 1 nm。12H 逐渐靠近16H,二者之间的距离逐渐缩短到0.098 0 nm,形成过渡态TS-4。随着反应越过过渡态,10C-14O新键生成,键长为0.136 5 nm,得到产物GBL;同时14O-16H 和10C-12H 键断裂,12H-16H 单键生成,脱除了另一份H2。
3.2.4 3-丁烯-1-醇异构化生成反-2-丁烯-1-醇
反应4 为BTO 经过过渡态TS-5 异构生成T-2BT1O 的反应。反应开始时,7C-8H 键长从0.109 9 伸长到0.179 8 nm,8H 向1C 方向移动;H 原子上电子主要向4C 上转移,8H 上的正电荷由原来的0.248 6增加到0.428 7;同时,1C-4C 键也随着8H 原子的移动开始从双键向单键的转变;4C-7C 单键也在逐渐缩短,键长从原来的0.150 5 减小到0.144 1 nm。8H 原子的移动使得分子上形成了一个1C-4C-7C-8H 的四元环,也标志着过渡态TS-5 的生成。当反应越过过渡态后,随着8H 向1C 方向的移动,7C-8H 键断裂,1C-8H 键逐渐形成。此时也伴随着双键从1C-4C 上移动至4C-7C 上。随着1C-8H 的键长缩短到0.109 4 nm,1C-4C 键长伸长到0.150 2 nm,4C-7C 键长缩短至0.133 9 nm,TS-5 转化为T-2BT1O。
3.2.5 3-丁烯-1-醇异构化生成顺-2-丁烯-1-醇
反应5 与反应4 的过程非常相似,是由BTO 经过过渡态TS-6 异构生成C-2BT1O 的反应,过程需要克服340.0 kJ·mol-1的势垒。反应开始时,9H 向1C 方向移动,同时1C-4C 键长逐渐增加而4C-7C 单键逐渐缩短,形成过渡态TS-6。当反应越过过渡态后,9H 与1C 成键,7C-9H 键断裂,双键从1C-4C 上转移到4C-6C 上,生成了C-2BT1O。
3.2.6 3-丁烯-1-醇脱水生成1,3-丁二烯
反应6 的机理与反应1 非常相近,BTO 克服277.5 kJ·mol-1势垒形成过渡态TS-7,随后脱水生成BDE。13O 原子首先向9H 原子靠近;H 原子上的电子主要向邻近的O 原子和C 原子上转移,9H 上的正电荷由原来的0.248 6 增加到0.443 6;同时,7C 原子上的负电荷由原来的-0.507 6 增加到-0.596 7。10C-13O 键以及7C-9H 键随着13O 向9H 方向的移动,其键长分别从0.143 0 以及0.109 9 增加到0.180 3 和0.148 2 nm。9H 原子的移动使得分子上形成了一个7C-10C-13O-9H 的四元环过渡态。当反应越过过渡态后,7C 上的电子继续向10C 上移动,同时9H 上的部分电子转移到13O 上形成新的化学键。7C 上的负电荷从-0.596 7 减少到-0.248 3,10C 上的负电荷也从-0.154 3 增加到-0.408 3。同时,随着9H 与13O 的成键,10C-13O以及7C-9H 键分别断裂,形成了H2O。7C 与10C 的键长由过渡态的0.143 4 nm 进一步缩短到0.134 3 nm,形成了C=C。至此,BTO 经过TS-7 脱水生成BDE。
3.2.7 3-丁烯-1-醇加氢生成丁醇
反应7 中,BTO 与H2克服353.4 kJ·mol-1的势垒形成过渡态TS-8 并进一步生成BuOH。反应开始时,17H、18H 原子彼此远离并分别向1C、4C 靠近;1C-4C 双键由原来的0.133 6 逐渐伸长到0.143 0 nm,并与17H 和18H 形成了一个四元环过渡态。其中17H 上的电荷为-0.225 9,而18H 上的电荷为0.392 6,可以认为反应的过程中,氢键先发生断裂,随后由18H 进攻1C,逐渐形成1C-18H 键;随即17H与4C 结合,形成过渡态TS-8。随后,17H-18H 断裂,1C-4C 由双键转变为单键,键长由0.143 0 增加到0.153 3 nm,最终生成产物BuOH,完成反应。
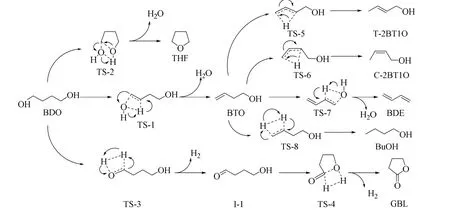
图9 BDO 反应网络中各反应的反应机理Fig.9 Mechanism of each elementary reaction in BDO reaction network
基于上述讨论的结果,BDO 反应网络中各基元反应的反应机理可以概括如图9所示。
3.3 1,4-丁二醇脱水反应网络的动力学分析
在得到各反应过渡态及相应的反应路径后,可以通过变分过渡态理论计算各反应经过隧道效应校正后的反应动力学常数,其结果如表2 所示。各反应的反应速率常数与反应温度的关系如图10 所示,通过双参数Arrhenius 方程进行拟合,可以得到各反应的指前因子和活化能[37],结果列于表2。
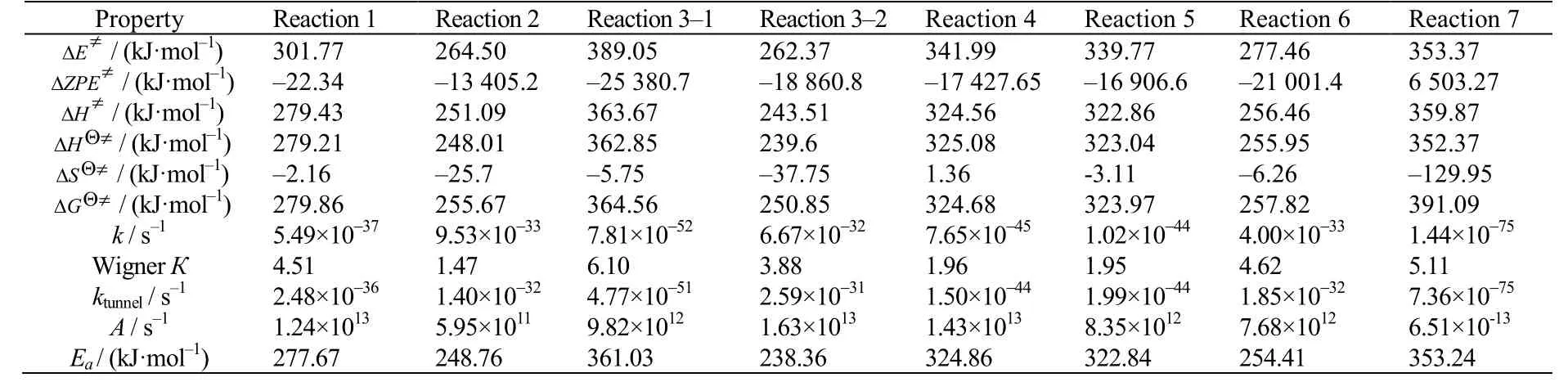
表2 过渡态热力学参数及各反应的动力学参数Table 2 Thermodynamic parameters of transition state and kinetic parameters of reactions
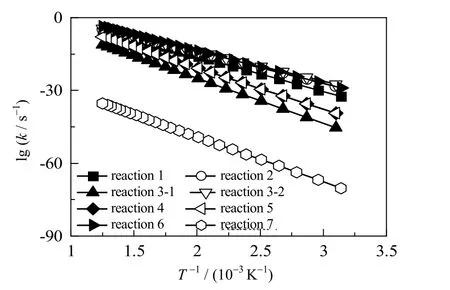
图10 不同温度下各反应的反应速率常数Fig.10 Rate constant of reactions at different temperatures
对BDO 脱水生成BTO 及两个平行副反应来说,反应1 的过渡态TS-1 势垒为301.8 kJ·mol-1,反应2 中过渡态TS-2 的势垒为264.5 kJ·mol-1。BDO 脱氢反应为两步反应,其中过渡态TS-3 的势垒为389.1 kJ·mol-1,过渡态TS-4 的势垒为444.8 kJ·mol-1。由于存在较高的势垒和较长的反应路径,BDO 脱氢反应显然比脱水反应更难发生。而从2 种BDO 脱水方式来看,BDO 脱水生成THF 反应的势垒相对较低,BDO 更容易环化脱水生成THF,其反应速率比反应1 高出4 个数量级。对于BTO 的串联副反应,则存在4 条平行的反应途径。BTO 异构生成T-2BT1O和C-2BT1O 的反应较为相似,反应能垒分别为342.0 和340.0 kJ·mol-1。反应生成BuOH 的势垒较高,为353.4 kJ·mol-1,该过程较难进行。而BTO 脱水生成BDE的势垒只有277.5 kJ·mol-1,反应容易进行。从各反应的反应路径中可以看出,脱氢和加氢反应所需的势垒最高,为反应网络中最难以进行的反应。BTO 双键异构化反应所需的势垒同样较高。而脱水反应所需的势垒则相对较低。其中,BDO 脱水生成THF 的反应以及BTO 脱水生成BDE 的反应势垒最低,对BTO选择性的影响最大。
通过上述分析可以发现,对于目标产物BTO 选择性影响最大的反应分别是BDO 脱水生成THF 的平行副反应2 以及BTO 进一步脱水生成BDE 的串联副反应6。分析反应1 和反应2 的反应机理可以发现,在脱水的过程中均首先涉及BDO 端羟基中14O 原子与相连1C 原子间C─O 键的断裂。在反应2 中,端羟基C─O 断裂后,羟基会进一步吸引另一个端羟基中的15H 原子,同时1C 原子也会进攻羟基中的13O原子,从而形成THF。反应2 的上述进程与Madduluri 等[38]的报道是一致的,这也说明本文中有关反应机理的计算分析结果是可靠的。
基于以上分析,在设计催化剂时,使用酸性催化剂对其羟基进行活化,使1C-14O 键更容易断裂,形成的碳正离子进攻另一端羟基中的O 原子,即可促使BDO 脱水环化生成THF。对于反应1 而言,在14O-1C 键断裂后,还需要断开β-H 原子与4C 原子之间的C─H 键,才能进一步脱水生成烯烃。通过对各原子上的电荷密度进行分析后可以发现,TS-1 中14O 上的负电荷为-0.745 9,5H 原子则带正电,其上电荷为0.218 1。基于此,应该设计一种酸碱双功能催化剂,以能够接受电子的酸性位点来活化羟基,同时采用可以给出电子的碱性位点对氢原子进行活化,二者协同作用才能使BDO 脱水生成BTO。
对于同样势垒较低的反应6 而言,其反应机理与反应1 非常相近,很难通过控制催化剂表面的活性位点来降低BDE 的生成。因此,可以采取提高反应空速,降低BTO 在催化剂床层中的停留时间来降低串联副反应的选择性,以提高目标产物BTO 的收率。
4 结 论
(1) 热力学研究表明:BDO 脱水反应网络中反应1、3、6 为吸热反应,其余反应为放热反应。升高温度有利于BTO 的生成。所有反应的Gibbs 自由能变均小于零,说明其在热力学上都是能够自发进行的。同时,当反应温度大于500 K 时,几乎所有反应的平衡常数ln KΘ都大于9.2,表明各个反应中逆反应发生的几率非常低,产物的分布主要取决于各反应的速率,过程为动力学控制。
(2) 通过对各反应进行过渡态搜索和IRC 计算,研究了各反应的过渡态结构、反应中键长及电荷的变化情况,提出了相应的反应机理。其中BDO 脱氢生成GBL 的反应为两步连续反应,其余反应均为一步反应。进一步的动力学研究表明,脱氢和加氢反应所需的势垒最高,反应最难进行。脱水反应所需的势垒则相对较低。其中反应2 和反应6 的反应势垒最低,对目标产物BTO 选择性的影响最大。
(3) 通过分析反应1 和反应2 的反应机理可以发现,两者均涉及BDO 端羟基C─O 键的断裂。而对于反应1,还需要经过β-H 原子与4C 原子之间的C─H 键断裂才能生成BTO。因此,可以通过设计酸碱双功能催化剂,以能够接受电子的酸性位点和可以给出电子的碱性位点分别对羟基和氢原子进行活化,促进BDO 脱水生成BTO。对于同样势垒较低的反应6,由于其反应机理与反应1 相似,难以通过控制催化剂活性位点来抑制BDE 的生成。因此,可以采取提高反应空速,降低BTO 在催化剂床层中的停留时间来提高目标产物BTO 的选择性。
符号说明:
A — 指前因子,s-1
E — 电子能量,kJ·mol-1
Ea— 活化能,kJ·mol-1
G — Gibbs 自由能,kJ·mol-1
H — 焓,kJ·mol-1
h — Planck 常数,6.63×10-34J·s
K — 反应平衡常数
k — 反应速率常数,s-1
kB— Boltzmann 常数,1.380649 × 10-23J·K-1
Q≠(T)、QR(T)
— 过渡态和反应物的单位体积配分函数
S — 熵,J·mol-1·K-1
s≠— 为变分过渡态位置
T — 温度,K
V — 势能,kJ·mol-1
ZPE — 零点能,kJ·mol-1
κ — 隧道校正因子
σ — 反应对称数
ω — 虚频频率,cm-1
ν — 化学计量系数
上标
s — 反应物数
n — 产物数
≠ — 过渡态结构
Θ — 标准状态
下标
m — 摩尔
r — 反应
R — 反应物
P — 产物
tunnel — 考虑隧道效应
i, j — 组分
