蛹虫草中新化合物的分离纯化、结构鉴定及抗肿瘤活性
2021-05-19刘健华王超然沈建福
项 婷,夏 琛,刘健华,王超然,沈建福,*
(1.浙江大学生物系统工程与食品科学学院,天然产物与人类健康研究中心,浙江 杭州 310058;2.浙江晟泰茶油科技有限公司,浙江 常山 324200;3.中国科学院大连化学物理研究所,辽宁 大连 116023;4.中科院大化所中国医药城生物医药创新研究院,江苏 泰州 225300)
蛹虫草(Cordyceps militaris(L.) Link),又名北虫草,是蛹虫草菌侵染昆虫蛹或接种在人工培养基上所形成的子实体,隶属于子囊菌门(Ascomycota)、虫草属(Cordyceps),是一种具有广泛食药用价值的真菌[1]。早在1986年,中国科学院就已经实现了蛹虫草的人工培育,与资源稀缺、产量低、价格高昂的野生型冬虫夏草相比,蛹虫草不仅克服了地域限制,而且同样含有多糖、有机酸、蛋白质、核苷、甾醇、无机元素、维生素等多种化学成分,具有抑菌、抗病毒、抗肿瘤[2]、免疫调节、神经保护、增强抗氧化活性、改善肠道损伤[3]等多种生物学功效,因此,人工培育的蛹虫草已经逐渐成为野生型冬虫夏草优良的替代品[4],应用开发前景极为广泛。
蛹虫草中最具有代表性的成分是核苷类物质,根据碱基不同,可将其分为嘌呤核苷和嘧啶核苷两大类,母核分别为胞嘧啶、尿嘧啶、胸腺嘧啶以及腺嘌呤、鸟嘌呤等[5]。朱丽娜等[6]运用高效液相分析法建立了蛹虫草中16 种核苷类成分的指纹图谱,包括胞嘧啶、尿嘧啶、胞苷、次黄嘌呤、黄嘌呤、尿苷、胸腺嘧啶、腺嘌呤、肌苷、鸟苷、脱氧鸟苷、胸苷、腺苷、虫草素、2′-脱氧腺苷和N6-(2-羟乙基)腺苷。大量研究表明,腺苷及其衍生物具有镇静、改善睡眠质量[7]、改善血脂和氧化还原能力、改善慢性肾脏疾病[8]、缓解神经炎症[9]、抗菌[10]、神经保护[11]以及广谱的抗肿瘤作用[12-16]。迄今,针对蛹虫草中核苷类物质的研究主要围绕虫草素和腺苷展开,而对于其他核苷类衍生物,例如氨基酸核苷衍生物的研究则较少,因此,深入挖掘蛹虫草中此类核苷衍生物,对于丰富蛹虫草中核苷类物质的数据库具有重要意义。
目前,分离纯化蛹虫草中核苷类物质的方法主要有大孔吸附树脂法[17]、离子交换树脂法、硅胶柱层析法[18]和制备液相色谱分离技术[19]等。离子交换树脂法操作过程繁琐;硅胶柱层析法吸附容量和回收率较低,并且需要大量的有机溶剂[20];大孔吸附树脂色谱法虽然价格低廉、稳定性高、易于再生,但是通常难以一步制备即达到较高纯度,需要与其他纯化方法联合使用。上述传统的分离方法,柱效低、分离能力差,适用于前处理、粗分离以及少数化合物的获得。制备液相色谱具有较强的分离能力,可根据物质的性质和液相色谱分析方法选择合适的柱填料和分离制备的方法,且能实现结构较类似的天然产物分离,但对于分子质量小、极性大的核苷类化合物而言,在反相色谱中保留很弱,是液相色谱分离分析中的难点。
在研究过程中发现,普通C18液相色谱柱对腺苷和虫草素的保留与分离较好,但在死时间附近,仍有不少成分共流出。为研究这类“不保留”的物质,本实验采用了一种极性共聚键合的色谱填料[21],利用其耐纯水的特性和增强的极性选择性,成功实现了多个化合物的保留与分离,其中包括虫草素、腺苷以及两种单体化合物。利用超高效液相色谱-四极杆飞行时间质谱(ultra-high performance liquid chromatography-quadrupole time-offlight mass spectrometry,UPLC-QTOF MS)、核磁共振波谱(nuclear magnetic resonance spectroscopy,NMR)法对所得的两种单体化合物进行结构鉴定,并通过噻唑蓝(methylthiazolyldiphenyl-tetrazolium bromide,MTT)法比较虫草素和两种单体化合物的抗肿瘤活性。
1 材料与方法
1.1 材料与试剂
蛹虫草 杭州中非生物科技有限公司。
虫草素标准品(纯度≥95.5%)、腺苷标准品(纯度≥99.7%) 中国食品药品检定研究院;三氟乙酸(色谱级,纯度≥99.5%) 上海阿拉丁生化科技股份有限公司;甲醇(HPLC级,99.9%) 美国TEDIA有限公司;醋酸(纯度≥99.5%) 国药集团化学试剂有限公司;有机溶剂均为国产分析纯。
HepG2肝癌细胞购自于中国科学院上海细胞库。
基本培养基(DMEM/High Glucose,含丙酮酸钠)康宁公司;胎牛血清(南美胎牛血清) 美国Gibco公司;胰酶(含乙二胺四乙酸)、磷酸盐缓冲溶液(phosphate buffer saline,PBS) 杭州科易生物技术有限公司;青霉素-链霉素混合液、二甲基亚砜(dimethyl sulfoxide,DMSO)、MTT溶液(5 mg/mL) 北京索莱宝科技有限公司。
1.2 仪器与设备
Alliance高效液相色谱仪(配有e2695高效液相色谱泵和二极管阵列检测器) 沃特世科技(上海)有限公司;Newstyle NP7000 SERIALS制备液相色谱(配有高压输液泵Newstyle NP7000 SERIALS PUMP和紫外-可见光检测器Newstyle NU3000 SERIALS UV/VIS DETECTOR,UV 260 nm) 江苏汉邦科技有限公司;C18分析型色谱柱(4.6 mm×2.5 mm,5 μm,100 Å)、XCharge C18分析型色谱柱(4.6 mm×2.5 mm,5 μm,100 Å)、C18-HCE制备型色谱柱(50 mm×250 mm,10 μm)、C18制备型色谱柱(50 mm×230 mm,10 μm)北京华谱新创科技有限公司;超高效液相色谱-四极杆飞行时间质谱联用仪 安捷伦科技(中国)有限公司;600兆超导核磁共振光谱仪 布鲁克(北京)科技有限公司;Forma 3111型二氧化碳细胞培养箱(37 ℃、5% CO2)美国赛默飞世尔科技有限公司;生物超净工作台 苏州苏净安泰有限公司。
1.3 方法
1.3.1 蛹虫草中活性物质的提取、分离与纯化
1.3.1.1 蛹虫草粗提液的制备
参考于戈等[22]的方法,并根据预实验的结果作适当修改。蛹虫草经真空冷冻干燥机干燥48 h,用粉碎机粉碎,过40 目筛,制成蛹虫草粉末,-18 ℃避光密封储存,备用。准确称取蛹虫草冻干粉末127.74 g,按照料液比1∶30(g/mL)加入体积分数为1%的醋酸溶液,40 ℃恒温水浴,搅拌提取40 min,真空抽滤,收集滤液和滤渣。滤渣进行二次提取,合并2 次提取所得滤液,减压浓缩,最终得到蛹虫草粗提液。
1.3.1.2 蛹虫草粗提液的分离制备
参考Zhang Yan等[23]的方法,对蛹虫草粗提液进行一维和二维分离制备。
一维制备:C18制备柱(50 mm×230 mm,10 μm),流速60 mL/min,上样量50 mL,甲醇-超纯水(15∶85,V/V),等度洗脱,据谱图出峰情况,收集各馏分,减压浓缩,得到一维制备各馏分的浓缩液。
二维制备:C18-HCE制备柱(50 mm×250 mm,10 μm),流速50 mL/min,洗脱时间0~40 min,甲醇0%~90%,超纯水100%~10%,梯度洗脱,据谱图出峰情况,收集各馏分,减压浓缩,得到二维制备各馏分的浓缩液。
1.3.2 高效液相色谱分析蛹虫草粗提液及其纯化组分
1.3.2.1 虫草素-腺苷混合标准曲线的建立
参照农业农村部标准规定的方法[24],并适当修改。准确称取虫草素和腺苷标准品各3.00 mg,分别用甲醇-水(含0.5% CF3COOH)(5∶95,V/V)溶解并定容至25 mL,作为虫草素和腺苷的标准储备液,质量浓度为120 μg/mL,4 ℃保存,有效期1 个月。分别取等体积腺苷和虫草素的标准储备溶液互溶,进行梯度稀释,得到腺苷-虫草素混合标准溶液,按1.3.2.2节中的方法进行测定。以腺苷-虫草素混合标准溶液质量浓度(μg/mL)为X轴,峰面积为Y轴,绘制腺苷-虫草素混合标准曲线。回归方程分别为:腺苷Y=57 279X-13 421,R2=0.999 9;虫草素Y=52 660X-4 281,R2=1,结果表明:腺苷和虫草素的质量浓度在1.875~120 μg/mL范围内均呈良好的线性关系。
1.3.2.2 高效液相色谱分析[24]
样品处理:取蛹虫草粗提浓缩液、二维制备各馏分浓缩液、腺苷-虫草素系列梯度的混标溶液各1 mL,过0.45 μm滤膜,用于高效液相色谱分析。
色谱条件:Alliance高效液相色谱仪;色谱柱:C18(4.6 mm×2.5 mm,5 μm,100 Å)、XCharge C18(4.6 mm×2.5 mm,5 μm,100 Å);流速:0.6 mL/min;紫外检测器波长260 nm;柱温:(35±5)℃;进样量:10 μL;洗脱条件:0~30 min,甲醇0%~10%,超纯水(0.5% CF3COOH)100%~90%,梯度洗脱。
测定:按照保留时间进行定性分析,样品与标准品保留时间的相对偏差不大于2%。按照标准曲线计算样品粗提溶液中腺苷、虫草素等物质的含量及最终得率。
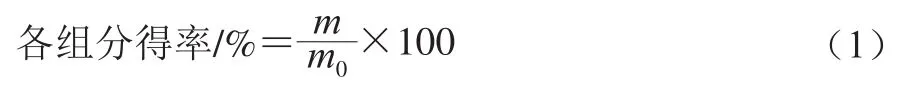
式中:m为各纯化组分冻干粉末的质量/g;m0为蛹虫草样品粉末的质量/g。
1.3.3 结构鉴定
对分离纯化得到的两种单体化合物(即图2a中5、6号峰),利用UPLC-Q-TOF MS、NMR法并结合相关的文献确定其化学结构式。
1.3.4 MTT法测细胞活力
采用MTT法检测各个组分对HepG2肝癌细胞活力的影响,参照文献[25]方法测定,细胞相对活力的计算方法如式(2)所示:
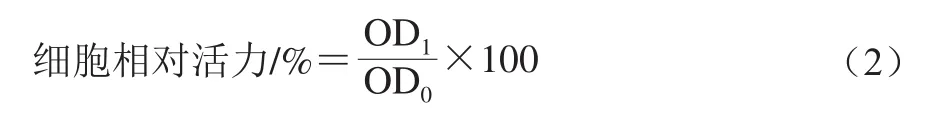
式中:OD1为样品组的吸光度;OD0为空白对照组的吸光度。
1.4 数据处理
2 结果与分析
2.1 高效液相色谱分析蛹虫草的粗提物
分别使用C18和C18-HCE两种液相分析型色谱柱,分析蛹虫草粗提液和腺苷-虫草素混合标准溶液,根据色谱结果选择对各组分分离效果较好的色谱柱作为后续实验的分析柱,结果如图1、2所示。
由图1a可知,在260 nm条件下,蛹虫草粗提液中分离度较大的组分仅有3 种,出峰时间主要集中在10~25 min,对照图1b可知其中两种组分分别为腺苷和虫草素,而样品中的其他成分的出峰时间大多集中在5 min左右,保留明显较弱,难以实现有效的分离纯化。

图1 C18色谱柱的高效液相色谱图Fig. 1 HPLC chromatograms on C18 column
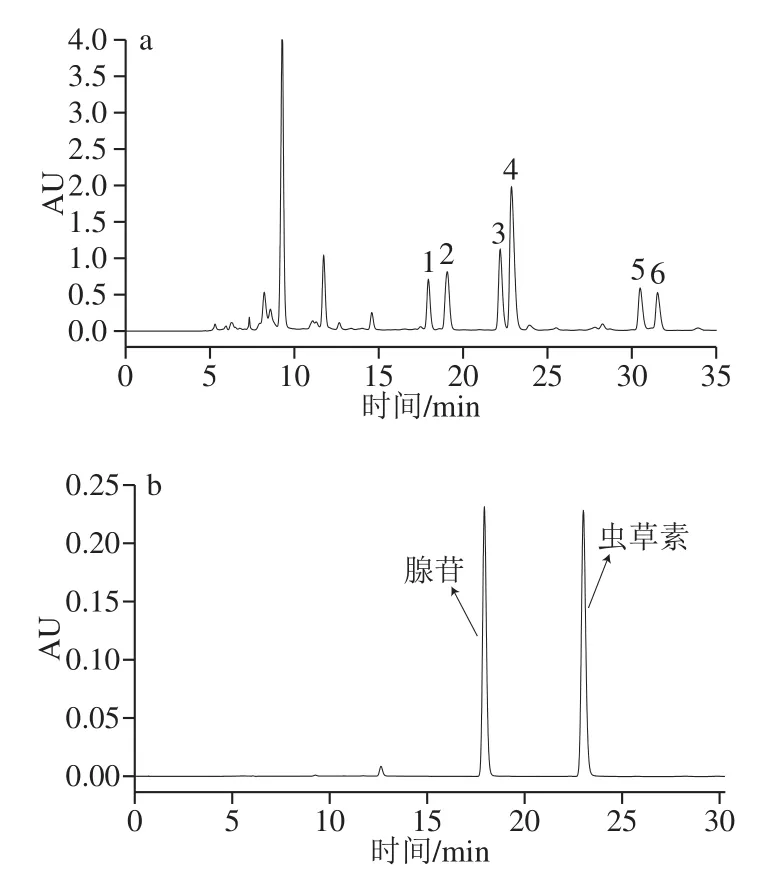
图2 C18-HCE色谱柱的高效液相色谱图Fig. 2 HPLC chromatograms on C18-HCE column
而在C18-HCE分析型液相色谱柱上(图2),蛹虫草粗提液中的成分能够较均匀分布在16~33 min的保留时间范围内,且可清晰识别6 个含量较高的色谱峰,对照标准品可知1、4号峰分别为腺苷和虫草素,有5 个色谱峰保留均比腺苷强,由此可见,C18-HCE分析型液相色谱柱对蛹虫草粗提液中各组分具有较好的选择性和分离效果[26]。经高效液相色谱法验证,5、6号峰即为普通C18分析型液相色谱柱中“不保留”的物质。
2.2 高效液相色谱分析分离制备所得的化合物
蛹虫草粗提液先后经C18和C18-HCE制备型液相色谱柱分离制备,得到了除腺苷和虫草素以外的2 种未知化合物,经高效液相色谱验证,确证两者即为图2a中的5、6号峰,暂且将其命名为化合物I和化合物II。根据各组分的高效液相色谱图(图3)可知,腺苷、虫草素、化合物I和化合物II的出峰时间分别为17.737、22.850、30.013 min和31.075 min。在260 nm条件下,4 种纯化组分的HPLC纯度分别为90.49%、98.11%、96.34%和98.33%,经冷冻干燥后获得冻干粉末,根据式(1)计算各组分得率分别为0.053%、0.253%、0.368%和0.231%。采用核磁共振技术进一步对化合物I和化合物II进行结构鉴定。

图3 经过二维制备后的4 种纯化组分在260 nm波长处的高效液相色谱图Fig. 3 HPLC chromatograms of the two purified components from two-dimensional preparation at 260 nm
2.3 未知单体化合物的结构鉴定
通过UPLC-QTOF MS、NMR并结合相关文献确定化合物I和化合物II的结构。
化合物I,是一种浅黄色粉末,经正、负离子模式UPLC-QTOF MS测定,m/z438.217 3[M+H]+(计算值438.213 5)、m/z436.204 4[M-H]+(计算值436.213 5),得出分子式为C17H27N9O5,不饱和度为9。由图4A可知,1H-NMR(600 MHz,DMSO-D6)δ8.51(s, 1H),8.42(s, 1H),8.23(s, 2H),8.15(s, 1H),7.34(s, 2H),5.97(m, 2H),5.41(s, 2H),4.52(m, 2H),4.02(s, 1H),3.72(dd,J=12.2, 2.1 Hz, 1H),3.61(t,J=6.0 Hz, 1H),3.53(dd,J=12.3, 4.1 Hz, 1H),2.93(dd,J=12.6, 6.5 Hz, 2H),1.64(m, 2H),1.35(m, 2H),1.29(m, 2H)。由图4B可知,13C-NMR(151 MHz,DMSO)δ171.42、158.80、156.12、152.60、148.87、139.12、119.14、89.46、83.10、73.19、60.94、53.02、50.56、40.06、32.19、29.76、22.04。
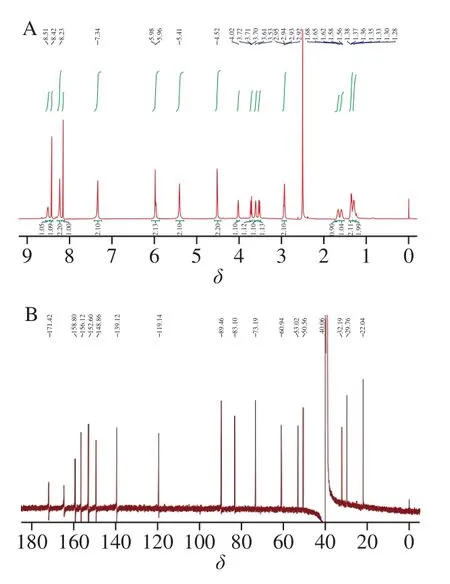
图4 化合物I的1H-NMR(A)和13C-NMR(B)谱图Fig. 4 1H-NMR (A) and 13C-NMR (B) spectra of compound I
化合物II:是一种深黄色粉末,经正、负离子模式UPLC-QTOF MS测定,m/z423.223 7 [M-NH+2H]+(计算值423.234 3)、m/z300.044 9 [MC5H2N5-H]+(计算值300.190 7),得出分子式为C17H28N10O4,不饱和度为9。由图5A可知,1H-NMR(600 MHz,DMSO)δ8.53(s, 1H),8.42(s, 2H),8.30(s, 3H),8.15(s, 1H),7.54(s, 3H),7.34(s, 2H),5.97(s, 1H),4.51(m, 1H),4.49(m, 1H),4.01(s, 1H),3.71(dd,J=12.2, 2.1 Hz, 1H),3.53(dd,J=12.3, 4.1 Hz, 1H),3.47(m, 1H),3.05(s, 2H),1.67(dd,J=13.5, 6.4 Hz, 1H),1.52(d,J=8.3 Hz, 1H),1.46(d,J=4.3 Hz, 2H),1.36(m, 2H)。由图5B可知,13C-NMR(151 MHz,DMSO)δ173.10、157.19、156.12、152.60、148.86、139.15、119.14、89.51、83.29、73.20、61.01、53.35、50.42、40.06、32.82、27.96、22.06。

图5 化合物II的1H-NMR(A)和13C-NMR(B)谱图Fig. 5 1H-NMR (A) and 13C-NMR (B) spectra of compound II
13C-NMR和无畸变极化转移增强波谱数据表明化合物I、II均有17 个碳信号,包括5 个亚甲基、5 个次甲基、2 个亚乙烯基和5 个季碳,唯一的差别在于化合物I比化合物II多1 个羰基碳。比较腺苷、虫草素、化合物I和化合物II的1H-NMR数据(表1),发现化合物I、II的部分核磁数据与腺苷、虫草素均存在高度相似。由于虫草素为3′-脱氧腺苷,是一种腺苷类似物,因此,推断化合物I、II与腺苷也存在相同母核。为此,比较腺苷与化合物I、II的13C-NMR数据,发现化合物I、II的类腺苷骨架部分(C1~C10)与腺苷几乎完全吻合,除了C6,这证明化合物I、II与腺苷确实存在相同母核。对比分析表2数据,化合物IδC6 50.56和化合物IIδC6 50.42相比于腺苷δC6 70.71均向高场位移,表明化合物I、II的类腺苷骨架部分在C6部位被电负性更小的取代基所取代[27]。
除了类腺苷骨架部分,化合物I、II其余的1H-NMR和13C-NMR数据均由取代的侧链产生,经分析比对发现侧链部分的13C-NMR数据与已知化合物高瓜氨酸[28]基本相同,说明化合物I、II类腺苷骨架C6取代侧链与其具有相同的碳骨架。再结合1H-1H COSY、NOESY以及HMBC远程相关等二维核磁数据,确定相邻碳上的氢在空间上的位置关系,最终确定化学结构式,发现两者皆为氨基酸腺苷衍生物,氨基酸作为侧链取代了腺苷骨架C6位上的羟基,如图6所示。

表1 化合物I、II与已知化合物的1H-NMR数据Table 1 1H-NMR data of compounds I, II and known compounds

表2 化合物I、II与已知化合物的13C-NMR数据Table 2 13C-NMR data of compounds I, II and known compounds

图6 化合物I(A)和化合物II(B)的结构式Fig. 6 Structural formulae of compound I (A) and compound II (B)
根据化学结构式,将化合物I和化合物II分别命名为2-amino-N-((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-2-(hydroxymethyl)tetrahydrofuran-3-yl)-6-ureidohexanamide,以及2-amino-N-((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-2-(hydroxymethyl)tetrahydrofuran-3-yl)-6-guanidinohexanamide。经SciFinder数据库检索比对,发现化合物II是一种全新的物质,尚鲜见有关文献报道该结构,它在蛹虫草生长过程中的合成途径以及生物学功效仍有待进一步研究。目前,对于化合物I的研究也较少,仅Lichtenthaler等[29]在1979年从蛹虫草中分离得到过该物质,囿于其在天然蛹虫草中含量有限,研究人员以高瓜氨酸和3′-氨基-3′-脱氧腺苷为原料,建立了一种人工合成化合物I的途径,并在生物学评价中证明该物质能够在肽链延长阶段干扰蛋白的合成,除此之外,尚鲜见其他文献报道化合物I的生物学活性。
2.4 各组分对HepG2肝癌细胞活力的影响

图7 不同质量浓度的虫草素和2 种化合物对HepG2肝癌细胞的相对活力的影响Fig. 7 Effects of different concentrations of cordycepin and the two compounds on the relative viability of HepG2 hepatoma cells
蛹虫草中的虫草素作为一种潜在的抗肿瘤药物被广泛关注,美国国家癌症研究所早在1997年就把虫草素引入18 种抗癌新药进行开发研究,大量实验表明虫草素能够通过抑制肿瘤细胞DNA合成[30]、调控肿瘤细胞周期[31-33]、直接诱导肿瘤细胞凋亡[34]等方式发挥其抗肿瘤作用。因此,本研究以虫草素为对照,采用MTT法,比较化合物I、化合物II和虫草素3 者对HepG2肝癌细胞作用的差异,其结果如图7所示。
由图7可知,经过上述3 种药物处理HepG2肝癌细胞后,HepG2肝癌细胞的相对活力随药物质量浓度的增大基本呈显著降低的趋势(P<0.05),且除化合物II以外,其他2 种药物对HepG2肝癌细胞的增殖抑制作用均随着作用时间的延长而明显增强。当质量浓度同为10 μg/mL的虫草素、化合物I和化合物II处理细胞24 h后,HepG2肝癌细胞的相对活力分别为(94.66±5.82)%、(95.34±8.77)%、(95.28±4.71)%,当质量浓度同为320 μg/mL的虫草素、化合物I和化合物II处理细胞24 h后,HepG2肝癌细胞的相对活力分别为(46.37±1.77)%、(39.22±1.36)%、(64.71±8.72)%,后者较前者HepG2肝癌细胞的相对活力降低了48.29%、56.13%、30.57%。同理可得,当作用时间为48 h时,HepG2肝癌细胞的相对活力分别降低了43.07%、61.57%、40.14%;当作用时间为72 h时,分别降低了51.35%、66.32%、43.32%。

表3 各组分对HepG2肝癌细胞的IC50Table 3 Half maximal inhibitory concentrations of cordycepin and the two compounds on HepG2 liver cancer cells
经分析计算得到3 种药物对HepG2肝癌细胞的IC50,如表3所示。结果表明,3 种药物均能抑制HepG2肝癌细胞的增殖,且浓度越大,作用时间越长,抑制效果越明显,抑制作用效果的顺序依次为:化合物I>虫草素>化合物II。长链脂肪酸衍生物被鉴定为一类新的具有抗MDA-MB-231乳腺癌活性的潜在药物,Koolaji等[35]通过制备多种结构类似物,确定了其中的活性基团。因此,可通过药物的构效关系分析本实验结果:化合物I中C6位氨基酸衍生物的取代侧链可能具有一定的生物活性,导致其对HepG2肝癌细胞的抑制作用强于虫草素;而化合物I与化合物II具有完全相同的碳骨架,属于结构类似物,两者在C17位上分别由羰基和氨基取代,羰基与氨基在疏水性、亲水性及空间位阻等方面的差异,可能影响药物的渗透性以及药物与靶点受体的结合能力,从而导致化合物I和化合物II对HepG2肝癌细胞抑制效果的不同。
3 结 论
C18和C18-HCE高效液相制备型色谱柱联用,能够有效改善核苷类化合物的保留和选择性,蛹虫草粗提液经过该方法分离制备后,得到腺苷、虫草素、化合物I和化合物II 4 种组分,其纯度和得率分别为90.49%、98.11%、96.34%、98.33%和0.053%、0.253%、0.368%、0.231%。经UPLC-QTOF MS、NMR鉴定,确定化合物I和化合物II均为氨基酸及其衍生物取代的核苷类衍生物,将两者分别命名为2-amino-N-((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-2-(hydroxymethyl)tetrahydrofuran-3-yl)-6-ureidohexanamid和2-amino-N-((2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-2-(hydroxymethyl)tetrahydrofuran-3-yl)-6-guanidinohexanamide,经过SciFinder数据库检索比对,确认化合物II是一种新化合物,尚鲜见有关文献报道,其在蛹虫草中的合成途径以及生物学功效仍有待进一步研究。比较虫草素、化合物I和化合物II的抗肿瘤活性,发现这3 种组分均能显著抑制HepG2肝癌细胞的增殖,抗肿瘤效果顺序依次为化合物I>虫草素>化合物II。药物抗肿瘤作用效果的差异可能与其结构相关,后续可针对药物结构的差异进行更深入研究,以确定活性基团,这对于新药合成,提高药效具有一定的参考价值。除此之外,本实验结果也为蛹虫草中氨基酸核苷类衍生物的分离纯化、结构鉴定、抗肿瘤作用的活性评价提供参考。
