The role of immune checkpoint inhibitors in triplenegative breast cancer: recent developments and future perspectives
2021-05-11MariosPapadimitriouZoiLiakouliChristosPapadimitriou
Marios Papadimitriou, Zoi Liakouli, Christos A. Papadimitriou
1Oncology Unit, Second Department of Surgery, Aretaieion University Hospital, National and Kapodistrian University of Athens,Medical School, Athens 11528, Greece.
2Radiotherapy Unit, First Department of Radiology, Aretaieion University Hospital, National and Kapodistrian University of Athens, Medical School, Athens 11528, Greece.
Abstract Triple-negative breast cancer (TNBC) represents the subtype of breast cancer with the most aggressive biological behavior and the worst prognosis compared to other breast cancers. Metastatic TNBC is characterized by a high proliferative index, rapid progression with metastases to the viscera and central nervous system, and generally an unfavorable prognosis with a survival of about one year. It is, therefore, necessary to identify specific targets and more effective treatments for patients with TNBC. Evidence of the effect of the tumor immune microenvironment on clinical outcomes is considered a significant issue in breast cancer therapeutics. Compared to other subtypes of breast cancer, TNBC is characterized by a higher mutational burden and is recognized as the most immunogenic among them. Based on these findings, immune checkpoint inhibition was evaluated in TNBC with encouraging results. Indeed, enhancing antitumor immunity in TNBC by blocking the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) axis or the programmed cell death-1 (PD-1) receptor/programmed death-ligand 1 (PD-L1) pathway is a promising treatment option. In this review, we examine the role of monoclonal antibodies targeting CTLA-4 and PD-1/PD-L1 in this breast cancer subtype and discuss combination approaches for early and advanced disease.
Keywords: Triple-negative breast cancer, immune checkpoint inhibition, CTLA-4, PD-1, PD-L1
INTRODUCTION
Triple negative breast cancer (TNBC) is a specific subtype of breast cancer which is immunohistochemically characterized by the lack of expression of estrogen receptors, progesterone receptors (PR), and HER2[1-5]. It represents about 15%-20% of all breast cancers and is a group of miscellaneous tumors with different molecular and clinicopathological characteristics that lead to different clinical behaviors. Although the majority of TNBCs are of basal-like phenotype and the majority of basal-like tumors (characterized by the frequent expression of cytokeratins 5/6 and the prominent membranous expression of EGFR) are TNBCs,clinical, microarray, and immunohistochemical data show these two terms are not synonymous. More specifically, the basal-like cancers are associated with a microarray gene-expression profile similar to that of basal or myoepithelial cells of the breast and represent the most homogeneous of all subtypes in the transcriptional term. TNBC, on the other hand, is routinely defined by immunohistochemistry and refers to a heterogeneous entity comprising several subtypes with distinct molecular characteristics. There is a discordance of up to 30% in the overlap between molecular and immunohistochemical definitions when comparing TNBC and basal-like cancers[1,3,4].
Distant metastases are more likely to develop in TNBC than in other subtypes of breast cancer with the risk peaking early at three years after initial diagnosis, while most deaths occur within the first five years, i.e.,TNBCs have a shorter median time to relapse and death. A large single-institution cohort study showed that patients with TNBC were significantly more likely to have died compared to those with other breast cancer subtypes (42.2%vs. 28%)[6]. The median time to death was 4.2 years for TNBC patientsvs. 6 years for patients with other breast cancers, and all deaths due to breast cancer in TNBC patients occurred within the first decade of diagnosis. On the other hand, disease cause-specific deaths in patients with luminal breast cancers continued to increase for up to 18 years after diagnosis. In addition, the rate of metastases was higher in patients with TNBC compared to other breast cancer subtypes (33.9%vs. 20.4%). Liet al.[7],reviewing SEER data for patients diagnosed with breast cancer between 2010 and 2012, found that patients with TNBC had worse overall survival and disease cause-specific survival than patients with other breast cancer subtypes at every stage.
Conventional chemotherapy still plays a central role in the treatment of metastatic TNBC (mTNBC) but with suboptimal results. The median overall survival (OS) in advanced/metastatic TNBC is approximately 12 months, an interval significantly shorter than the corresponding seen in other breast cancer subtypes[5-7].Therefore, identifying specific molecular targets and consequently more effective/personalized therapies for TNBC patients remains a significant clinical challenge. Among the most promising treatments is immunotherapy for reasons related to the characteristics of the disease that may make it more responsive to this type of therapy. One of these features is a higher tumor mutational burden (TMB), since TNBC has been shown to have more median mutations compared with other breast cancer subtypes. High TMB could increase T-cell responses against a wide-spectrum of tumor-specific neoantigens. In TNBC, a positive correlation was found between TMB and tumor immune cell infiltration, while tumor infiltrating lymphocytes (TILs) have prognostic value and are associated with improved clinical outcomes in this tumor entity[8,9].
In this article, we look at the role of immune checkpoint inhibitors in the treatment of TNBC and review the most important completed clinical trials, as well as those in progress.
MOLECULAR AND IMMUNOPHENOTYPIC CHARACTERIZATION OF TNBC
Due to its aggressive biological behavior and the heterogeneity which characterizes tumors within the TNBC group, the molecular classification of these neoplasms is an important research priority for the better identification of molecular-targeted therapies. Thus far, several groups have investigated the biology of TNBC using a variety of tools, such as immunohistochemistry, gene expression analysis, and sequencing.Lehmannet al.[10], analyzed TNBC gene expression profiles obtained from 21 breast cancer datasets and identified 6 different TNBC subtypes. The two basal-like subtypes (BL1 and BL2) were heavily enriched in cell cycle pathways and DNA damage response genes; the immunomodulatory subtype (IM) was enriched for genes whose expression regulates immune cell processes; the mesenchymal and mesenchymal stem-like subtypes had a high expression of genes involved in epithelial-mesenchymal transition and growth factor pathways; and, finally, the luminal androgen receptor (LAR) subtype was characterized by a prominent androgen receptor signaling. In a more recent study from the Baylor University[11], the RNA and DNA profiles of 198 TNBCs were analyzed, which led to the identification of four subtypes: LAR, mesenchymal,basal-like immune-suppressed (BLIS), and basal-like immune-activated (BLIA). BLIS and BLIA cancers had the best and worst prognosis, respectively, in terms of disease-free survival (DFS). The researchers also identified many highly expressed molecules that could possibly represent targets for more effective treatment of TNBC. Finally, researchers from France[12]separated TNBCs by a fuzzy clustering method into three clusters. Twenty-two percent of patients had not basal-like tumors, enriched in luminal subtypes and positive androgen receptor (C1), 45% were pure basal-like (C2), and 33% of them had high immune responses (C3). In contrast to the studies from Vanderbilt and Baylor, the French study did not recognize a mesenchymal subtype. However, despite the various methodologies used in the aforementioned studies and the different number of identified subtypes, it is consistently suggested that TNBC is comprising of four subtypes: basal-like, mesenchymal, luminal androgen receptor, and immune-enriched[13].
A special genetic feature of TNBC is its association with germlineBRCAmutations. Indeed, TNBC patients have a higher incidence of germlineBRCA1/2mutations, with a rate of approximately 15%-20% according to various studies[14-17]. BRCA1 and BRCA2 proteins are key components of the homologous recombination(HR) pathway and cells lacking these proteins are unable to repair double-strand breaks (DSBs) by HR.Because functionalBRCA1andBRCA2genes are involved in maintaining genome stability, their inactivation may lead to oncogenic transformation and further progression to breast cancer. Furthermore,this genomic instability gives rise to breast cancers with increased likelihood of immunogenic somatic mutations (and consequently of neoantigens), that is cancers with an immunogenic phenotype[18,19]. In a whole-genome sequences analysis of 560 breast cancers, 90 had germline or somatic inactivating mutations inBRCA1orBRCA2or showedBRCA1promoter methylation. These cancers carried many genomic rearrangements[20]. Wen and Leong[19]evaluated data from two breast cancer datasets (The Cancer Genome Atlas and the Wellcome Sanger Institute) and found thatBRCA1-andBRCA2-deficient breast cancers had significantly higher numbers of mutations when compared toBRCA1/2-proficient tumors. Furthermore,BRCA1-deficient breast cancers were associated with increased expression of PD-L1 and PD-1, a significantly greater number of immune cells infiltrating the tumor microenvironment, and an enriched T cell-inflamed gene expression profile when compared toBRCA1/2-proficient tumors. However, these findings were not observed inBRCA2-deficient tumors. In addition to the aforementioned study, other studies have also shown that TNBC is characterized by a higher TMB compared to luminal breast cancers that may lead to a higher incidence of immunogenic mutations[21,22]. Of note, TMB has been associated with both response to immunotherapy and improved clinical outcomes in patients with various malignancies[23,24].
In early-stage breast cancer, TIL count is associated with improved survival, decreased risk of distant recurrences, and an increased likelihood of responding to neoadjuvant chemotherapy (NACT)[25-27]. A systematic review of fifteen studies[28]showed that TNBC had the highest incidence of TILs, and their count may represent a potential biomarker for response to immunotherapy. TNBC has also a higher rate of PD-L1 expression when compared to the luminal subtypes[29,30], and this underlines the therapeutic role of monoclonal antibodies (moAbs) targeting the PD-1/PD-L1 axis. Recently, the FDA approved atezolizumab(anti-PD-L1 moAb) in combination with nab-paclitaxel for the treatment of PD-L1-positive advanced/metastatic TNBC[31].
IMMUNOTHERAPEUTIC APPROACHES IN TRIPLE-NEGATIVE BREAST CANCER
The immune system consists of two different parts, the innate (non-specific) and the adaptive (specific or acquired) immunity, which have overlapping functions. Innate immune cells, such as natural killer (NK)cells and cells of myeloid origin, act as an organized network against foreign (non-self) antigens without having been previously stimulated by them. These cells then activate the adaptive immunity through antigen presentation - which is one of their major functions - while enhancing their own activities, such as phagocytosis for macrophages and granulocytes and natural cytotoxicity for NK cells[32,33]. Functional responses of the adaptive immune system are achieved by its stimulation by professional antigen-presenting cells, such as dendritic cells (DCs). Adaptive immunity includes components of humoral immunity (B lymphocytes) and components of cell-mediated immunity (T lymphocytes). The main functions of the adaptive immune system include recognition of specific foreign antigens during the process of antigen presentation, generation of responses that are adapted to efficiently eliminate potential pathogens or pathogen-infected cells that escape innate host defenses, and development of immunologic memory[33,34].
The immune system plays a composite role in breast cancer pathogenesis and progression[35,36]. In the context of cancer biology, the various immune reactions that protect against cancer development are referred to as immunosurveillance.
However, despite the existence of functional immunosurveillance, individuals still develop cancers, which indicate the role of immunity not only in the elimination but also in the progression of breast cancer. This complex network of evolving interactions between breast cancer and host immunity is characterized by the term “immunoediting”, a process consisting of three phases: in the elimination phase, the immune system effectively targets and eliminates breast cancer; in the equilibrium phase, a balance is obtained between cancer progression and its elimination by the immune system; and, finally, in the escape phase, cancer persists and overcomes the host’s immunity, progressing and metastasizing to other organs[37]. Early in breast cancer oncogenesis, acute inflammation activates innate immunity, leading to tumor cell death and maturation of DCs that trigger a tumor-specific T-cell response. It is at this stage that either rejection of the tumor or selection of breast cancer cell populations capable of escaping the immune response will occur[36].The transition from an acute to a chronic inflammation phase establishes a composite tumor microenvironment (TME) comprising suppressive immune cells and stromal cells, such as fibroblasts and endothelial cells, which allow immune escape to occur and breast cancer to progress[38,39]. During this transition, the CD4+T-cell response deviates from T helper (Th) type 1 to Th type 2[40], immune checkpoint molecules are upregulated on both tumor and immune cells[41], and more importantly immune-suppressive metabolic pathways are activated in various immune cell types[42,43]. All these mechanisms together create a powerful network of immunosuppression within the breast tumor microenvironment.
Today, most immunotherapy strategies used in breast cancer are designed to bypass diminished immune stimulation. In their majority, these immunomodulatory approaches aim to enhance T-cell responses, either by targeting inhibitory pathways with immune checkpoint inhibitors or by targeting activating pathways. As in all malignancies, TNBC immunotherapy can be divided into passive and active. Passive immunotherapy includes the administration of immunomodulating agents and adoptive cell transfer, which enhance the existing antitumor immune response. Further, immunomodulating agents include co-stimulatory agonists,such as stimulatory moAbs to 4-1BB, OX40, and GITR and co-inhibitory antagonists, such as stimulation through checkpoint blockade [cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), PD-1, and PD-L1].Adoptive cell transfer refers to the transfer into a patient of autologous TILs, genetically engineered T-cell receptor (TCR) cells, or chimeric antigen receptor T cells. Active immunotherapy, on the other hand, aims to stimulate the immune system and is divided into specific (cancer vaccines) and non-specific (cytokines and immune adjuvants)[44-51].
There are currently more than 300 ongoing clinical trials evaluating the role of immunotherapy in breast cancer. Most of these trials focus on assessing immune checkpoint inhibitors alone or in various combinations in TNBC, with approximately 80% of studies being performed in the metastatic setting[52,53].One of the first trials with immune checkpoint inhibitor monotherapy in mTNBC was the phase Ib trial KEYNOTE-012 that assessed pembrolizumab in heavily pretreated patients[54]. In this trial, monotherapy with pembrolizumab achieved an overall response rate (ORR) of 18.5%. However, these results of singleagent pembrolizumab were considered discouraging when compared to those obtained in other solid tumors, such as melanoma and lung cancer[53]. Other early clinical trials of monotherapy with immune checkpoint inhibitors (atezolizumab or avelumab) in mTNBC demonstrated ORR of around 10% or less[55-57]. ORR with monotherapy also appears to be lower in PD-L1-negative baseline patients, in patients with ≥ 2 prior chemotherapy lines for their metastatic disease, and in patients with prior exposure to taxanes[52]. In most studies, immune checkpoint inhibitors were combined with chemotherapy, other inhibitors, or biologic agents. The suggestion to combine immune checkpoint inhibitors with chemotherapy was based on the observation that chemotherapeutic agents and other types of treatment (including irradiation, photodynamic therapy, or cryoablation) could promote the so-called immunogenic cell death(ICD). This particular type of cell death is the result of the activation of adaptive immune responses in dying cancer cells due to the exposure or secretion of immunostimulatory molecules[52,58,59].
CTLA-4 (CD152) INHIBITION
The binding of TCR to the cell surface proteins of the major histocompatibility complex (MHC) does provide specificity to T-cell activation, but other co-stimulatory signals are also needed. More specifically,the B7-1/B7-2:CD28/CTLA-4 co-stimulatory pathway plays a crucial role in regulating T-cell activation and tolerance. Binding of B7-1 (CD80) or B7-2 (CD86) molecules, which are expressed on professional APCs,with CD28 molecules on the T cells initiates and regulates T-cell response, a process involving T-cell proliferation, differentiation, and survival and characterized by increased energy metabolism and upregulation of various genes, such as cell survival genes[49,60,61].
CTLA-4 shows high affinity for B7-1/2 (CD80/86). The molecule is not constitutively expressed on T lymphocytes, but is induced following T-cell activation[49,50,60]. While the CD28:B7-1/2 binding acts as a costimulatory signal for T-cell proliferation and activation, CTLA-4:B7-1/2 interaction serves as a coinhibitory signal aimed at preventing early activation of Tcells[62].
CTLA-4 is recognized as the “leader” among the inhibitory immune checkpoint molecules, as it stops potentially autoreactive T cells at the initial stage of naïve T-cell activation. Binding of B7-1/2 molecules to CTLA-4 on CD8+T lymphocytes results in the dephosphorylation of TCR signaling proteins, among which CD3 leads to T-cell inhibition. Inhibitory signals produced by CTLA-4 binding to the B7-1/2 molecules counterbalance the stimulatory signals derived from CD28:B7-1/7 and TCR:MHC binding. Mechanisms of this opposing effect on T-cell response include capturing and removing B7-1/2 from the membrane of APCs, modulation of T-cell motility that leads to decreased interaction with APCs, and inhibition of TCR signal transduction by binding to TCRζ chain and consequently inhibiting tyrosine phosphorylation after Tcell activation[63-66].
In resting naïve T cells, CTLA-4 molecules are located mainly intracellularly. Stimulatory signals resulting from both TCR:MHC and CD28:B7-1/7 binding lead to increased expression of CTLA-4 molecules on the cell surface due to exocytosis of CTLA-4-containing vesicles. It is, therefore, a graded feedback loop where stronger TCR signaling causes more translocation of CTL-4 molecules to the cell surface[66,67]. Figure 1 illustrates CD80/86:CTLA-4 interactions and CTL-4 blockade with moAbs, while main molecular aspects of this pathway are shown in Figure 2. Inhibition of CTLA-4 with moAbs leads to increased activation of the immune system and represents a novel strategy in cancer therapeutics. Table 1 summarizes CTLA-4 inhibition strategies with moAbs in TNBC, in both neoadjuvant and metastatic setting.
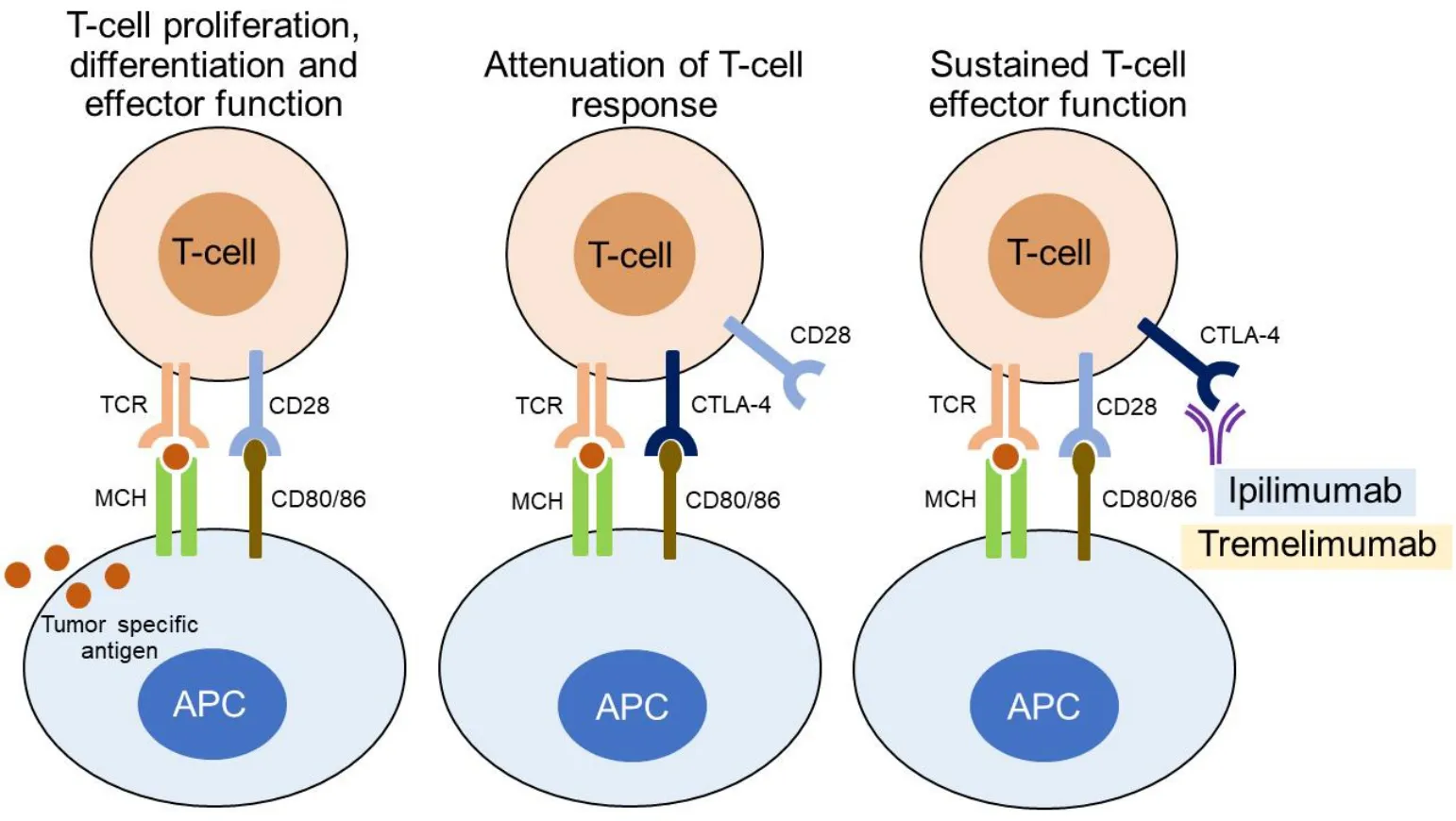
Figure 1. CD80/86:CTLA-4 interactions and CTL-4 blockade with monoclonal antibodies.

Figure 2. Main molecular aspects of the CD80/86:CTLA-4 pathway.
Monoclonal antibodies targeting CTLA-4
Ipilimumab
In 1995, Krummel and Allison[68]described the antagonist effect between CD28 and CTLA-4 on the response to T-cell stimulation. They generated an antibody to CTLA-4 (9H10) to investigate the effects of engagement of this molecule in a carefully defined system using highly purified T cells and concluded that the outcome of T-cell stimulation is regulated by CD28 costimulatory signals, as well as inhibitory signals derived from CTLA-4. One year later, Leachet al.[69]described that administration of anti-CTLA-4 antibodies resulted in the rejection of tumors in mouse models. Furthermore, tumor rejection induced by CTLA-4 blockade generated immunity that protected experimental animals at second exposure to cancer cells. The authors concluded that blocking the inhibitory effects of CTLA-4 could enable and enhance effective immune responses against cancer cells. Following these pioneering studies, several subsequent studies in animal experimental models have demonstrated that CTLA-4 blockade leads to regression of established tumors by increasing anticancer activity due to reversal of CD8+T-cell tolerance.
Ipilimumab, a fully human IgG1 moAB that blocks CTLA-4, improved overall survival when administered with or without a gp100 peptide vaccine in patients with metastatic melanoma[70]. The therapeutic indications of ipilimumab include metastatic melanoma (as monotherapy or in combination with nivolumab) and intermediate/poor-risk advanced renal cell carcinoma in combination with nivolumab as first-line treatment[71]. To date, no completed clinical trials with single-agent ipilimumab in TNBC have been published. There is an interesting clinical report for a patient with metastatic TNBC who was treated with low-dose immune checkpoint blockade (concurrent nivolumab and ipilimumab) with IL-2 and locoregional hyperthermia and whole-body hyperthermia. The treatment resulted in complete clinical remission[72]. Selected current clinical trials with the CTLA-4 inhibitor are presented in Table 2.
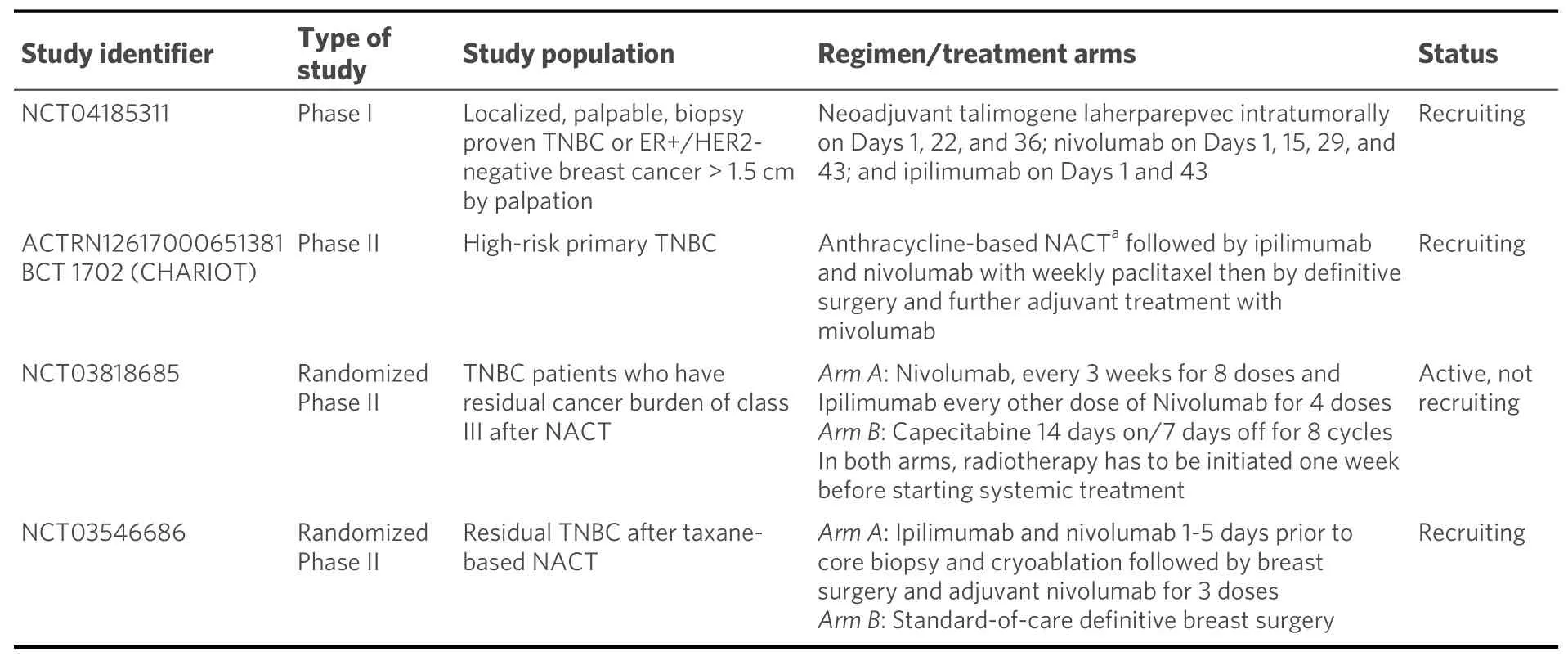
Table 2. Selected current clinical trials investigating ipilimumab in TNBC
1. Ipilimumab in the neoadjuvant setting
Because it has been demonstrated in murine models that intracellular cryopreservation with immune modulation generates a strong systemic anti-tumor immune response, it was thought that this approach could be applied to improve outcomes in early breast cancer. A pilot study investigated the safety and tolerability of preoperative, single-dose ipilimumab and/or cryoablation. T-cell activation was observed in the blood of patients receiving a single-dose of ipilimumab, while a moderate increase in the ratio of tumor CD8+Ki67+T cells to T regulatory (Treg) cells after combination therapy only was documented[73]. Based on these findings, a phase II trial has been designed in which 160 patients with operable TNBC and residual disease ≥ 1.0 cm after taxane-based NACT will be randomized to either breast surgery only (control arm) or ipilimumab/nivolumab/cryoablation followed by breast surgery and adjuvant nivolumab (intervention arm). Adjuvant capecitabine is recommended for all patients[74].
The phase II BCT 1702-CHARIOT trial has been designed for patients who have residual TNBC of at least 15 mm and less than 50% reduction in its longest diameter after four cycles of standard anthracycline-based NACT. The trial combines weekly paclitaxel for 12 doses with ipilimumab and nivolumab. Postoperatively,nivolumab is administered every four weeks for nine doses[75]. The aim of the CHARIOT trial is to test whether these patients with unfavorable prognosis could benefit from combined immune checkpointblockade plus chemotherapy.
2. Ipilimumab in mTNBC
The phase II study NIMBUS will assess the efficacy of nivolumab plus ipilimumab in patients with hypermutated (TMB ≥ 10 mutations/megabase) HER2-negative metastatic breast cancer and ≤ 3 prior lines of chemotherapy for advanced disease[76].
Tremelimumab
Tremelimumab is a fully human IgG2 moAb targeting CTLA-4[77-79]that binds to the same epitope with ipilimumab on the CTLA-4 molecule[80]. Although initial phase I and II trials of tremelimumab were promising[81-83], a phase III trial in patients with metastatic melanoma was discontinued after reviewing early interim data, which indicated that treatment with tremelimumab would not be superior to standard chemotherapy[84]. Following the presentation of the findings of this trial, interest in the development of tremelimumab in the therapeutics of various malignancies, including breast cancer, was significantly reduced. Recently, however, it is being tested again, mainly in combination with other immune checkpoint inhibitors.
1. Tremelimumab in mTNBC
A pilot study was designed to determine ORR and evaluate immunogenomic characteristics in 7 patients with metastatic TNBC and 11 patients with metastatic luminal breast cancer who received the combination of tremelimumab and durvalumab. All responses (3/7) were observed among patients with mTNBC.Responders showed an overexpression ofCD8,granzyme A, andperforin 1genes and a higher mutational and neoantigen load when compared to non-responders. Furthermore, patients with TNBC demonstrated an oligoclonal shift of the most abundant TCRβ clonotypes compared to those with luminal disease[85].
Based on the results of preclinical studies suggesting that ionizing radiation induces ICD, thus enhancing,through multiple mechanisms, the systemic antitumor immune response[86-90], a phase I study assessed the safety of tremelimumab on the third day of palliative radiotherapy in patients with metastatic breast cancer.Among the six patients enrolled, five had luminal disease and one had mTNBC. Tremelimumab in combination with radiotherapy appeared to be a tolerable treatment, while an increase of proliferating Tregs in peripheral blood was observed one week after treatment in five patients[91].
Selected current clinical trials with tremelimumab in combination with moAbs targeting the PD-1/PD-L1 pathway are presented in Table 3.
PD-1/PD-L1 INHIBITION
ThePDCD1gene and the corresponding surface receptor PD-1 (CD279) were first discovered in a murine T-cell hybridoma and a murine hematopoietic progenitor cell line and was thought to induce cell death[92].However, it has since been clarified that PD-1 is primarily involved in inhibitory immune signaling and is an essential regulator of adaptive immune responses[49,66,93,94]. Although PD-1 is not expressed on most circulating T cells, its expression can be induced upon stimulation, through the TCR complex or exposure to cytokines[95,96]. Other cell types, such as B cells, mast cells, and myeloid dendritic cells, can also express PD-1 protein, which can regulate their own and bystander cell functions under pathophysiological conditions[97,98].
PD-1 binds to counter-receptors PD-L1 (B7-H1; CD274) and PD-L2 (B7-DC; CD273). PD-L1 is expressed on miscellaneous cell types, including tumor cells, APCs, infiltrating immune cells[99], and endothelial and epithelial cells[49,94]. PD-L1 expression on tumor cells is induced by inflammatory cytokines but may also be constitutively upregulated by autonomous cancer cell mutations or gene alterations. In T cells, binding of PD-1 to its ligand causes phosphorylation of tyrosine residues, initiating an intracellular downstream signaling event that transmits the dephosphorylation of TCR proximal signaling components. Among these,CD28 is the primary target, as the PD-1-associated SHP-2 preferentially dephosphorylates CD28 over the TCRζ chain and other substrates[100-106]. By intervening in the early stages of TCR/CD28 signaling and relative IL-2-dependent positive feedback, PD-1 signaling leads to decreased cytokine production, cell cycle progression, andBCL2L1gene expression, as well as decreased expression of transcription factors involved in T cell effector functions[105,106]. PD-1 activity is, therefore, only relevant when T cells are activated by TCRdependent signaling[107]. Overall, PD-1 pathway is vital for maintaining effective immunity and peripheral self-tolerance, so as to avoid autoimmunity and immunopathology[49,66,94]. Figure 3 illustrates PD-1/PD-L1 interactions and their blockade with moAbs, while main molecular aspects of the PD-1/PD-L1 signaling are shown in Figure 4. Neutralizing moAbs targeting the PD-1/PD-L1 pathway have resulted in dramatic improvements in the outcome of various advanced neoplasms. Table 4 summarizes PD-1/PD-L1 inhibition strategies with moAbs in TNBC.

Table 4. PD-1/PD-L1 inhibition strategies with monoclonal antibodies in the various settings of TNBC: single-agent treatment and combinations

Figure 3. PD-1/PD-L1 interactions and their blockade with monoclonal antibodies.

Figure 4. Main molecular aspects of the PD-1/PD-L1 pathway.

?
characterization of the immune profile of residual tumors.
In the context of the neoadjuvant setting, the ongoing phase II BELLINI trial evaluates the efficacy of nivolumab in combination with ipilimumab[113].
2. Nivolumab in mTNBC
A. Nivolumab with chemotherapy and/or irradiation
In metastatic TNBC, nivolumab has been assessed in combination with other agents. A phase I trial evaluates the safety of nivolumab with nab-paclitaxel in three cancer types: HER2-negative metastatic breast cancer, advanced NSCLC with carboplatin, and advanced pancreatic cancer with or without gemcitabine.Breast cancer patients are treated with two different dosing regimens of the combination of nivolumab with nab-paclitaxel. Eligibility criteria include 0-1 prior lines of chemotherapy in the metastatic setting and

?
The Dutch TONIC trial is of particular interest[118,119]. Because treatment with anti-PD(L)-1 moAbs benefits a small percentage of unselected patients with mTNBC[36,52,53], strategies have been proposed to make the TME more susceptible to PD-1/PD-L1 pathway inhibition. As existing preclinical data suggest immunomodulatory properties for both chemotherapy and irradiation[120-122], induction treatment with either low-dose chemotherapy or irradiation aimed at stimulating antitumor immune responses could represent such a strategy. In the first stage of the TONIC trial, 67 patients with mTNBC who had received ≤3 prior lines of chemotherapy were randomly allocated to nivolumab or to a two-week induction treatment followed by nivolumab. This induction treatment consisted of irradiation of one metastatic lesion; two weekly infusions of doxorubicin; oral cyclophosphamide; or two weekly infusions of cisplatin. After the induction period, all patients received nivolumab until disease progression by iRECIST criteria[123]. ORR was 20% in the entire population. Most responses were observed in the doxorubicin cohort (ORR 35%) followed by those in the cisplatin cohort (ORR 23%). After induction with doxorubicin or cisplatin, an upregulation of immune-related genes involved in PD-1/PD-L1 and T-cell cytotoxicity pathways was detected. This was further supported, after induction with doxorubicin, by enrichment among upregulated genes associated to inflammation, JAK-STAT and TNF-α signaling. All these clinical and translational data suggest that priming with short-term doxorubicin and cisplatin may create a more favorable TME, thus increasing the likelihood of response to immune checkpoint inhibitors in mTNBC[119].
B. Nivolumab in combination with other immune checkpoint inhibitors
Inducible T-cell costimulator (ICOS) or CD278, the third member of the CD28/CTLA4 family, is an immune checkpoint costimulatory molecule that is expressed on activated T cells playing a crucial role in cell signaling, immune responses, and regulation of cell proliferation. ICOS activity is considered important,especially for Th2 cells, to direct appropriate antibody responses to foreign antigens[124-128]. The ICOS axis has a dual effect by playing two opposite roles, and this depends on its expression on the various T-cell subtypes and the pathophysiological circumstance[128]. More specifically, it is an activator of antitumor T-cell responses, but also can participate in pro-tumor responses and immune invasion due to its close connection with the suppressive activity of Tregs. In preclinical models, ICOS agonist moAbs have been shown to potentiate the effect of immune checkpoint inhibition, whereas antagonist moAbs reduce immunosuppressive Tregs, while inhibiting ICOS-expressing lymphoid cells. Therefore, the therapeutic modification of the ICOS pathway should theoretically be based on the administration of either an antagonist or an agonist moAb, depending on the characteristics of the tumor and its microenvironment[129-131].
JTX-2011 is an agonist moAb that binds to and activates ICOS. The phase I/II ICONIC trial[132]assessed the safety, tolerability, and preliminary efficacy of JTX-2011 alone and in combination with ipilimumab,nivolumab, or pembrolizumab in patients with various advanced and/or refractory types of cancer,including TNBC. JTX-2011 monotherapy and combined treatment with nivolumab were well tolerated and showed antitumor activity in heavily pre-treated patients with mTNBC[133].
C. Nivolumab in combination with molecular-targeted agents
A phase II study is assessing the efficacy of the combination of nivolumab plus cabozantinib in patients with mTNBC who have received ≤ 3 previous lines of chemotherapy for their metastatic disease[134]. Cabozantinib is a tyrosine kinase inhibitor (TKI) that strongly binds to and inhibits several tyrosine kinases, such as c-MET (hepatocyte growth factor receptor, HGFR), vascular endothelial growth factor receptor types 1-3(VEGFR-1/2/3), c-KIT, FLT-3, TIE-2, tropomyosin-related kinase B (TRKB), and AXL[135-139]. The design of this study was based on the assumption that cabozantinib may enhance the activity of nivolumab by reducing myeloid-derived suppressor cells, while increasing the number of T cells. Peripheral blood samples and tumor biopsies will be taken before the start of treatment, during it, and at disease progression for immune profiling and for examining biomarkers of the response to treatment.
D. Nivolumab with immunomodulating cytokines
Interleukin-2 (IL-2) is a pleiotropic cytokine with a broad spectrum of activities including functions leading to proliferation and survival of T cells. IL-2 also enhances the cytolytic activity of CD8+T cells and NK cells,induces the differentiation of Tregs, and mediates activation-induced cell death[140,141]. Treatment of metastatic melanoma and renal cell carcinoma with IL-2 at high doses was approved by FDA in the 1990s,providing up to 25% objective responses[142-145]. However, high-dose IL-2 has not been widely used due to severe toxicity associated with the overactivation of the immune system[145].
Bempegaldesleukin (BEMPEG or NKTR-214) is a prodrug in clinical development comprising IL-2 protein bound by multiple releasable polyethylene glycol (PEG) chains. This agent was developed to harness the immunostimulatory benefits of the IL-2 pathway, maximize anticancer responses, and minimize AEs. When bempegaldesleukin is administeredin vivo, PEG chains are released slowly, creating a cascade of increasingly active IL-2 conjugates bound by fewer PEG chains. The PEG chains on bempegaldesleukin are located in the region of IL-2 that contacts the α subunit of the heterotrimeric IL-2 receptor complex,IL2Rαβγ, thus reducing bempegaldesleukin’s ability to bind to and activate the heterotrimer. Because the IL2Rαβγ complex is constitutively expressed on Tregs, PEGylation reduces the cytokine for IL2Rαβγ to a greater extent than for IL2Rβγ, which is the predominant receptor complex on CD8+T cells. It has been shown in multiple syngeneic models that treatment with bempegaldesleukin favors the activation of CD8+cells over Tregs in the TME providing antitumor activity[146,147].
The PIVOT-02 phase I/II trial[148]is investigating the efficacy and safety of bempegaldesleukin in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, NSCLC,urothelial carcinoma, or TNBC. The dose escalation stage enrolled 38 patients and established the recommended phase II dose. The expansion phase is evaluating the combination in patients who are either immune therapy naïve or have relapsed/refractory disease to prior PD-1/PD-L1 inhibition. Among patients with TNBC, who were included in the trial and evaluated for response, ORR was 13%, regardless of PD-L1 expression, and DCR 45%. ORR was 11% in patients with PD-L1-negative tumors and 17% in PD-L1-positive patients. ORR was 20% in patients with ≥ 2 lines of prior therapy, regardless of PD-L1 expression.Adverse events of Grade ≥ 3 were observed in 23% of patients and included dehydration, hypotension, and myalgia. The predictive and prognostic factors assessed in PIVOT-02 trial included PD-L1 status, age, DFS,LDH, number and type of metastatic sites, and prior taxane-based chemotherapy.
Interleukin-10 (IL-10) is considered a prototypical anti-inflammatory cytokine, which contributes significantly to the maintenance and restoration of immune homeostasis[149-151]. Recent evidence, however,suggests that the cytokine has pleiotropic roles, such as supporting B-cell and CD8+T-cell activation[151].Experimental data in mouse models show that IL-10 can inhibit the development of metastases[152], while additional data support the immunostimulatory capacity of IL-10 in an immune-oncology context[153-155].However, the short biological half-life of IL-10in vivorestricts its therapeutic application because it requires large and frequent dosage administration[156,157]. PEGylation of IL-10 leads to the product pegilodecakin,providing increased serum half-life and prolonged systemic exposure, also allowing one subcutaneous injection of pegilodecakin daily[157,158]. Pegilodecakin monotherapy has shown activity in patients with advanced cancers, and PEGylated cytokine alone or in combination with anti-PD-1 treatment leads to proliferation and expansion of PD-1+ Lag-3+CD8+T cells and expansion of novel CD8+T-cell clones[159]. In a phase Ib trial, patients with various malignancies refractory to previous therapies (among them one woman with TNBC) were treated with pegilodecakin combined with nivolumab or pembrolizumab[160].
Pembrolizumab
Pembrolizumab is a humanized IgG4 kappa moAB directed against human cell surface PD-1[108,109,161,162].Pembrolizumab is indicated for the treatment of a wide spectrum of malignancies, including melanoma,metastatic NSCLC (alone or in combination with chemotherapy), squamous cell carcinoma of the head and neck, urothelial carcinoma, gastric cancer, cervical cancer, Merkel cell carcinoma, dMMR metastatic solid tumors, hepatocellular carcinoma, and Hodgkin’s disease[163,164].
As previously mentioned, the phase Ib KEYNOTE-012 study was one of the first immune checkpoint inhibitor monotherapy trials[54]. Although its results have been considered discouraging, regarding mTNBC,more recent trials that evaluated the combination of pembrolizumab with other agents have yielded promising results. Selected current clinical trials with pembrolizumab alone or in combination with other agents in TNBC are presented in Table 6.
1. Pembrolizumab in the neoadjuvant setting: combinations with chemotherapy
In the context of neoadjuvant therapy, the results of three very interesting trials were published in the first half of 2020. The phase Ib KEYNOTE-173 study assessed the efficacy and safety of NACT plus pembrolizumab in high-risk, early-stage TNBC[165]. Six different pembrolizumab plus chemotherapy regimens were evaluated. All patients were given pembrolizumab, every three weeks, for a total of nine cycles. In Cycle 1, patients received pembrolizumab monotherapy, while, in Cycles 2-5, they also received one of the following chemotherapeutic regimens: weekly nab-paclitaxel alone (Cohort A); weekly nabpaclitaxel plus carboplatin every three weeks (Cohort B); weekly nab-paclitaxel plus carboplatin every three weeks (Cohort C); nab-paclitaxel plus carboplatin, both weekly (Cohort D); weekly paclitaxel plus carboplatin every three weeks (Cohort E); and paclitaxel plus carboplatin, both weekly (Cohort F). All patients were also given, in Cycles 6-9, doxorubicin plus cyclophosphamide, every three weeks. The incorporation of carboplatin into the nab-paclitaxel/paclitaxel regimen (Cohorts E and F) was performed,because previous studies have shown that the adding carboplatin and/or bevacizumab to NACT with taxane and anthracycline significantly increases the rate of pathological complete responses (pCR), in stage II/III TNBC[166,167]. Response to NACT, in the sense of pCR, predicts an improved OS in TNBC[168].
In KEYNOTE-173, the pCR rate (ypT0/Tis ypN0) was 60% across all cohorts. This rate was the same with and without carboplatin, but it was higher for patients who received carboplatin every three weeks compared with those who were treated with weekly carboplatin (63%vs. 55%). Overall, the highest pCR rates were obtained with regimens that combined weekly nab-paclitaxel and carboplatin (Cohorts B-D), but they were associated with higher toxicity and, therefore, did not meet the predefined recommended phase II dose threshold. Event-free survival (EFS) rates at 12 months were 100% for patients who achieved ypT0/Tis ypN0 responsevs. 88% for those who did not, while OS rates at 12 months were 98% for patients who received carboplatinvs. 80% for patients who did not. Higher pre-treatment PD-L1 combined positive score(CPS) and pre- and on-treatment stromal TILs were significantly associated with higher pCR rates.Immune-related AEs and infusion reactions occurred in 30% of patients and were of Grade ≥ 3 in 10% of them.

?

?
In the aforementioned KEYNOTE-012 trial[54], pembrolizumab was administered to patients with advanced/metastatic TNBC, gastric cancer, urothelial cancer, and head and neck cancer that expressed PDL1 in the stroma or in ≥ 1% of tumor cells by immunohistochemistry (PD-L1-positive tumors). Among TNBC patients, ORR was 18.5% with a median time to response of 17.9 weeks. The median duration of response was not yet reached at the time of analysis. Adverse events of Grade ≥ 3 were observed in 15.6% of patients including one treatment-related death.
The phase II study KEYNOTE-086 had two cohorts. Cohort A included 170 pre-treated women with mTNBC, regardless of PD-L1 expression[172], while Cohort B included 84 women with PD-L1-positive disease who had not previously received systemic treatment in the metastatic setting[173]. All patients in both cohorts were given pembrolizumab, every three weeks, for up to two years. In Cohort A, the ORR was 5.3%for the total population and 5.7% for the PD-L1-positive subpopulation. Median PFS and OS were 2.0 and 9.0 months, respectively. AEs of Grade ≥ 3 were observed in 12.9% of patients. In Cohort B, ORR amounted to 21.4% and DCR to 23.8%. Median PFS and median OS were 2.1 and 18.0 months, respectively. Eight patients (9.5%) experienced AEs of Grade 3, whereas AEs of Grade 4 or death were not observed.
Cortéset al.[174]presented at the 2019 ESMO Meeting the results of the KEYNOTE-119 study that randomized 622 patients with mTNBC in a 1:1 ratio to receive, as second- or third-line systemic treatment,pembrolizumab monotherapy or chemotherapy by investigator’s choice (including capecitabine, eribulin,vinorelbine, and gemcitabine). All patients had been previously exposed to anthracyclines and taxanes.Median OS (primary endpoint) did not differ statistically significantly between subpopulations with tumors with CPS ≥ 10 or CPS ≥ 1 compared with the overall population, although the therapeutic effect of pembrolizumab increased as CPS increased.
B. Pembrolizumab and irradiation
Radiotherapy is thought to enhance the immune response by promoting pro-immunogenic cell death and apoptosis, as well as priming and activating cytotoxic T cells[120,175,176]. Based on this rationale, which was the same as in the TONIC trial[119], a phase II study assessed the efficacy and safety of pembrolizumab plus radiotherapy in mTNBC, regardless of PD-L1 expression[177]. Patients received 3000 cGy of radiotherapy delivered in five daily fractions. The total dose of radiotherapy administered and its fractionation were based on the clinical feasibility and efficacy, in the context of palliative care, and preclinical data suggesting immunostimulatory benefits of hypofractionated doses in breast cancer[178]. Indications for radiotherapy included palliation of pain or control of metastases that developed while the patient was on systemic antitumor treatment. Pembrolizumab was given 1-3 days after the first fraction of radiotherapy and then every three weeks until disease progression was diagnosed or unacceptable toxicity occurred. The ORR for the entire population (17 patients) was 17.6%. All three patients who achieved CR also had complete disappearance of all lesions outside the irradiated field. The most common toxicity of Grade 1 or 2 was dermatitis (29%).
C. Pembrolizumab with chemotherapy
A phase Ib trial of pembrolizumab plus chemotherapy (PembroPlus) was performed to evaluate their safety and efficacy in 49 patients with various advanced/metastatic solid tumors, including 12 breast cancer patients. Pembrolizumab was administered with one of the following chemotherapy regimens: gemcitabine,gemcitabine plus docetaxel, gemcitabine plus nab-paclitaxel, gemcitabine plus vinorelbine or irinotecan,and liposomal doxorubicin. There were eight PRs across multiple tumor types[179].
An ongoing phase II trial is evaluating the combination of pembrolizumab with nab-paclitaxel in patients with metastatic HER2-negative breast cancer who have received ≤ 2 previous treatment lines for advanced/metastatic disease. In addition to clinical efficacy and safety, various parameters related to the effect of treatment on ΤΜΕ will be examined, as well as predictive factors of response to the therapeutic combination. These include mutational and neoantigen load, TILs, TCR by immunosequencing, and immune gene profiles in tumor samples. The predictive role of PD-L1 expression in tumor tissue will also be assessed, but the expression is not among the inclusion criteria. Finally, the possible role of the intestinal microbiome in modifying the immune response will be examined[180].
Another ongoing multicenter phase II study is evaluating pembrolizumab following a single priming dose of cyclophosphamide, in patients with advanced/metastatic TNBC. The primary endpoint is to estimate PFS,whereas the reduction of circulating Tregs represents the co-primary endpoint. Secondary endpoints include ORR, duration of response, DCR, and OS[181]. The rationale for this study was based on the wellestablished knowledge that low-dose cyclophosphamide has the capacity to deplete intratumoral Tregs in various tumors, including breast cancer[182-184]. Tregs can suppress antitumor immunity, thereby inhibiting effective immune responses in the host and hindering immunosurveillance of neoplasia, thus promoting tumor growth and progression[185]. Therefore, early infiltration of the TME with Tregs indicates a possible mechanism for the failure of immune checkpoint inhibition in certain tumor types which are heavily infiltrated by adaptive immune cells. Of note, infiltration with Tregs is significantly increased in TNBC and claudin-low breast cancer compared to other breast cancer subtypes[186,187]. Claudin-low tumors have been recognized to preferentially display a triple-negative phenotype and Treg depletion potentiates immune checkpoint inhibition in this subtype[187].
As previously mentioned in the context of ICD, the combination of chemotherapy with immune checkpoint inhibitors may enhance the activity of the latter[121,122]and this has been clearly demonstrated in lung cancer by combining platinum-based chemotherapy with immunotherapy[188-192]. An ongoing randomized phase II trial is enrolling patients with refractory breast cancer with chest wall disease. Patients are randomly allocated in a 2:1 ratio to either pembrolizumab plus carboplatin, every three weeks for at least six cycles,followed by single-agent pembrolizumab (Arm A), or carboplatin, every three weeks (Arm B), until disease progression. Cross over to pembrolizumab on disease progression is allowed for patients in the control arm(Arm B). The primary endpoint is to estimate DCR at 18 weeks of treatment. The secondary endpoints include toxicity, PFS, and ORR based on iRECIST criteria and PD-L1 expression[193].
The following two studies were conducted with the same rationale. A pilot phase II study assessed the efficacy and safety of concurrent pembrolizumab plus investigator-selected first- or second-line paclitaxel or oral capecitabine in mTNBC. Toxicities were generally consistent with monotherapy experience and improved with dose-reduction. ORRs were 43% and 25% for capecitabine plus pembrolizumab and paclitaxel plus pembrolizumab, respectively. Interestingly, patients enrolled < 12 months after treatment with curative intent had numerically lower responses than patients who did not progress rapidly. No significant differences in immunomodulation were observed between the two types of chemotherapy,however, both capecitabine and paclitaxel were associated with a decrease in T-cell count[194]. A phase II study conducted by Shahet al.[195]assessed the combination of pembrolizumab plus capecitabine administered in three-week cycles in mTNBC or endocrine-refractory metastatic luminal breast cancer. For the entire population, median OS was 15.4 months and median PFS 4.0 months, which was not significantly longer than that of three months in historical controls. The ORR and clinical benefit rate (CBR) were not significantly different between disease subtypes (ORR 13% and 14%, CBR 25%, and 29% for TNBC and endocrine-refractory luminal breast cancer, respectively). Of note, some patients experienced a prolonged disease control.
D. Pembrolizumab with molecular-targeted agents
Poly (ADP-ribose) polymerase (PARP) inhibitors target the DNA base excision repair (BER) pathway and the repair of DNA single strand breaks (SSBs). If PARP activity is inhibited and BER is impaired, unrepaired SSBs accumulate, and they degenerate in replication forks to become DSBs. DSBs are repaired by various repair pathways, with HR representing the most important of them. In the process of HR repair, BRCA1 and BRCA2 proteins play a central role. The concept of synthetic lethality by inhibiting PARP in HR defective tumors is thought to be due to failure to repair SSBs. Indeed, in the absence of HR, accumulating SSBs that degenerate to DSBs prove lethal as they persist, while they can only be repaired by alternative pathways, such as non-homologous end joining, that are prone to errors[5,196]. FDA has approved the PARP inhibitors olaparib and talazoparib for germlineBRCA1/2mutated metastatic breast cancer[197,198].
In the phase II study TOPACIO, 55 women with advanced or metastatic TNBC, regardless ofBRCA1/2mutation status or PD-L1 expression, were administered oral niraparib plus pembrolizumab. ORR was 21%and DCR 49%, respectively. In patients carryingBRCA1/2mutations, ORR was 47% and DCR 80%,respectively, with a median PFS of 8.3 months. Treatment was safe and warrants further investigation,especially in patients withBRCA1/2-mutated mTNBC[199].
BGB324 (bemcentinib) is an oral inhibitor of AXL, a cell surface receptor tyrosine kinase (RTK), part of the TAM family of kinases. All three TAM family RTKs are pleiotropic inhibitors of the innate immune response. AXL is expressed throughout all normal tissue and cell types and overexpressed in various malignancies including chronic and acute myelogenous leukemia, NSCLC, melanoma, gastric and colorectal cancer, breast cancer, and prostate cancer. Binding of AXL to its ligand GAS6, a vitamin K-dependent protein, leads to the activation of various signaling pathways, such as PI3K-AKT-mTOR, MAPK/ERK, NF-κ B, and JAK/STAT. In tumors, AXL activation regulates cellular pathways involved in cell survival,proliferation, apoptosis, epithelial-mesenchymal transition, cell adhesion, invasion, migration, and immune suppression[200-205]. Blocking AXL signaling with BGB324 has been shown to enhance the effect of immune checkpoint blockade in lung and mammary adenocarcinoma models[206]. A phase II study (BGBC007) is evaluating the efficacy of pembrolizumab plus BGB324 in patients with previously treated, advanced or metastatic TNBC[207].
E. Pembrolizumab with immunomodulating agents/cytokines
PGG (Imprime) is a novel β-1,3/1,6 glucan biologic derived fromSaccharomyces, which utilizes innate immune effector cells to enhance killing of antibody-targeted, complement-opsonized tumor cells. More specifically, when administered intravenously, endogenous anti-b glucan antibodies bind to PGG. Through the classical pathway of complement activation, PGG becomes opsonized and then binds to complement receptor 3 (CR3) on circulating neutrophils and monocytes. Thus, PGG modulates innate immune functions, leading to “priming” of these cells. In the TME, when a complement-activating anti-tumor moAb binds to tumor cell antigens, it induces complement deposition on the tumor cells. Subsequently,chemoattractant components produced in the complement cascade attract innate immune cells to the TME.Primed neutrophils and macrophages can exert anti-tumor activity against opsonized tumor cells through a CR3-dependent mechanism, while they can also modulate responses of other cells[208-210]. In a phase II study(IMPRIME 1), 44 previously treated patients with mTNBC received PGG plus pembrolizumab until PD or intolerable toxicity. Tumor biopsies and blood samples were also assessed for PGG-mediated immune activation. ORR was 15.9%, while an additional 38.6% of patients had SD as best response (DCR at any time 54.5%). Median OS was 13.7 months. Furthermore, confirmed responses were evident in patient subpopulations with poor prognosis, such as patients with high LDH levels, visceral metastases, and liver metastases. O'dayet al.[211]concluded that these clinical data suggest that PGG provides added clinical benefit to pembrolizumab.
Finally, a phase IIa trial is evaluating a chemokine modulating (CKM) pre-treatment followed by pembrolizumab in patients with mTNBC previously treated with at least one line of systemic therapy[212].Pre-clinicalex-vivodata show that a CKM regimen consisting of rintatolimod, IFNα, and celecoxib selectively attracts cytotoxic T cells into tumors and increases intratumoral expression of PD-1/PD-L1/PDL2, without enhancing soluble suppressive mechanisms[213]. Furthermore, experimental data from murine models demonstrate the safety of the combined CKM and PD-1 blockade, as well as efficiency in inducing long-term survival of mice with resistant tumors[212]. Rintatolimod, a mismatched double stranded polymer of RNA, is a restricted toll-like receptor 3 and represents a medication intended for treatment of chronic fatigue syndrome/myalgic encephalomyelitis[214].
F. Pembrolizumab in combination with antibody-drug conjugates
Ladiratuzumab vedotin (LV, SGN-LIV1A) is an antibody-drug conjugate targeting LIV-1 protein. LV consists of an IgG1 antibody conjugated through a proteolytically cleavable linker to monomethyl auristatin E, a potent microtubule-disrupting agent. LIV-1 is a transmembrane protein with zinc transporter and metalloproteinase activity, which is expressed in > 60% of mTNBCs. Its expression has been found to be associated with lymph node metastasis and metastatic progression. LV antitumor activity is thought to primarily be the result of intracellular payload release, leading to mitotic arrest and apoptotic cell death[215-219]. LV induced apoptosis is consistent with immunogenic cell death[218]. SGNLVA-002, a phase Ib/II trial of LV plus pembrolizumab as first-line treatment for patients with locally advanced/metastatic TNBC, is currently enrolling in the USA and EU[220].
G. Pembrolizumab with vaccines
SV-BR-1-GM is a vaccine consisting of irradiated allogeneic breast cancer cells, derived from the cell line SV-BR-1. These cells are transfected with theGM-CSFgene and secrete GM-CSF when administered intradermally, thus potentiating a tumor-specific cytotoxic T-lymphocyte immune response[221,222].
Α phase I/IIa trial assessed SV-BR1-GM in combination with immune checkpoint inhibitors in patients with advanced/metastatic breast cancer refractory to standard therapy[223]. Patients received intravenous cyclophosphamide prior to intradermal injection of SV-BR-1-GM and IFNα into the inoculation sites approximately two and four days later. Cycles were administered every two weeks × 3, and then monthly ×3. Immunologic responses were measured by delayed type hypersensitivity (DTH) after each inoculation. A similar regimen was used with SV-BR-1-GM in combination with pembrolizumab with cycles administered every three weeks. In the phase I/IIa trial, among 23 patients who underwent 1-8 cycles of treatment, tumor response was observed in three (13%) of them, all of whom matched SV-BR-1-GM at least at one HLA allele. There were no related serious AEs. A measurable DTH response was present in 21 patients. Of patients who developed a DTH response and had at least one HLA match, the tumor regression rate was 33%, and it was 67% for those with two HLA matches. In responders after treatment, blood lymphocytes showed increased cytokine secretion following stimulation with antigens expressed in SV-BR-1-GM.Twenty-one patients had expression of PD-L1 in identified circulating cancer-associated cells, and expression levels increased with treatment. Therefore, a combination study with pembrolizumab was initiated. Data on the first six patients show that the regimen is clinically active and safe. One patient with a robust DTH response had evidence of tumor regression in liver metastases. The study is ongoing and has been modified to evaluate the combination of PD-1 inhibitor INCMGA00012 with the IDO inhibitor epacadostat.
Spartalizumab
Spartalizumab (PDR001) is a humanized IgG4κ moAb that binds PD-1 with subnanomolar activityin vitro[224,225].
1. Spartalizumab in mTNBC
A. Spartalizumab monotherapy
In a phase I study, Japanese patients with advanced malignancies who progressed on standard therapy received spartalizumab in three dose groups. The most common cancer types were ovarian cancer (17%),head and neck cancer (11%), cervical cancer (11%), and TNBC (11%). Overall, ORR was 11% with partial responses seen in patients with hepatocellular carcinoma and transitional cell carcinoma[226].
B. Spartalizumab in combination with monoclonal antibodies targeting other immune checkpoint molecules
In a phase I/II trial, ieramilimab (LAG525) plus spartalizumab was dosed at 15 dose levels/schedules in patients with various advanced malignancies, including TNBC. The combination produced durable responses in two out of five women with mTNBC. Furthermore, treatment with LAG525 plus spartalizumab resulted in immune profile modulation, i.e., a trend to convert the biomarker profiles from immune-cold to immune-activated in TNBC tumor biopsies[227]. Ieramilimab is a humanized moAb directed against the inhibitory lymphocyte activation gene-3 (LAG-3) receptor. LAG-3 is expressed on activated CD4+and CD8+T cells, a subset of Tregs, NK cells, B cells, and plasmacytoid dendritic cells. This inhibitory receptor suppresses T-cell activation, proliferation, and homeostasis and has been reported to play a role in the suppressive function of Tregs[228-232]. Upon administration, ieramilimab binds to LAG-3 expressed on TILs and blocks its binding to MHC class II molecules that are expressed on tumor cells. Targeting LAG-3 with antagonistic moAbs may invigorate the immune response to cancer, while preclinical evidence supports promising synergy especially with simultaneous inhibition of the PD-1/PD-L1 pathway[229,230].
C. Spartalizumab in combination with immunomodulating agents
A phase Ib/II study assessed lacnotuzumab with spartalizumab in patients with advanced/metastatic melanoma, endometrial cancer, pancreatic cancer, or TNBC[233]. Lacnotuzumab (MCS110) is a high affinity humanized moAb targeting CSF-1 and inhibiting CSF-1/CSF-1R interactions. Inappropriate overexpression of CSF-1/CSF-1R has been documented in breast, prostate, and ovarian cancer, as well as in Hodgkin’s lymphoma. CSF-1R signaling was found to enhance the invasion and metastasis of solid tumors[234-237]. In malignant neoplasms, the increased activity of the CSF-1/CSF-1R axis leads to increased infiltration of the TME by TAMs which are important tumor-promoting cells, especially in breast cancer, stimulating tumor progression and mediating resistance to PD-1 inhibitors[231,235,238]. In the trial conducted by Calvoet al.[233],lacnotuzumab was combined with spartalizumab at six dose levels. By RECIST 1.1 criteria, there was one PR, while the SD rate was 19%. By iRECIST criteria, disease control rate was 27%. The combination of lacnotuzumab with spartalizumab was well tolerated overall.
ADU-S100 (MIW815) is a synthetic cyclic dinucleotide agonist (activator) of stimulator of interferon genes(STINGs), a receptor crucial to activate the innate immune system. ADU-S100 (MIW815) activates all known human and mouse STINGs and effectively induces the expression of cytokines and chemokines,leading to a robust and durable antigen-specific T-cell-mediated immune response against cancer cells[239-241]. In a phase Ib study, patients with metastatic cancers or lymphomas received intratumoral injections of ADU-S100 with intravenous spartalizumab at various doses/schedules[242]. No DLTs were reported during the first cycle at any dose level. The most common (≥ 5 patients) treatment-related AEs were pain at the injection site, pyrexia, and diarrhea. PRs were observed in TNBC patients without prior exposure to anti-PD-1 treatment and in melanoma patients with recurrent/refractory disease to prior anti-PD-1 treatment.
Selected current clinical trials with spartalizumab in combination with other agents in metastatic TNBC are presented in Table 7.
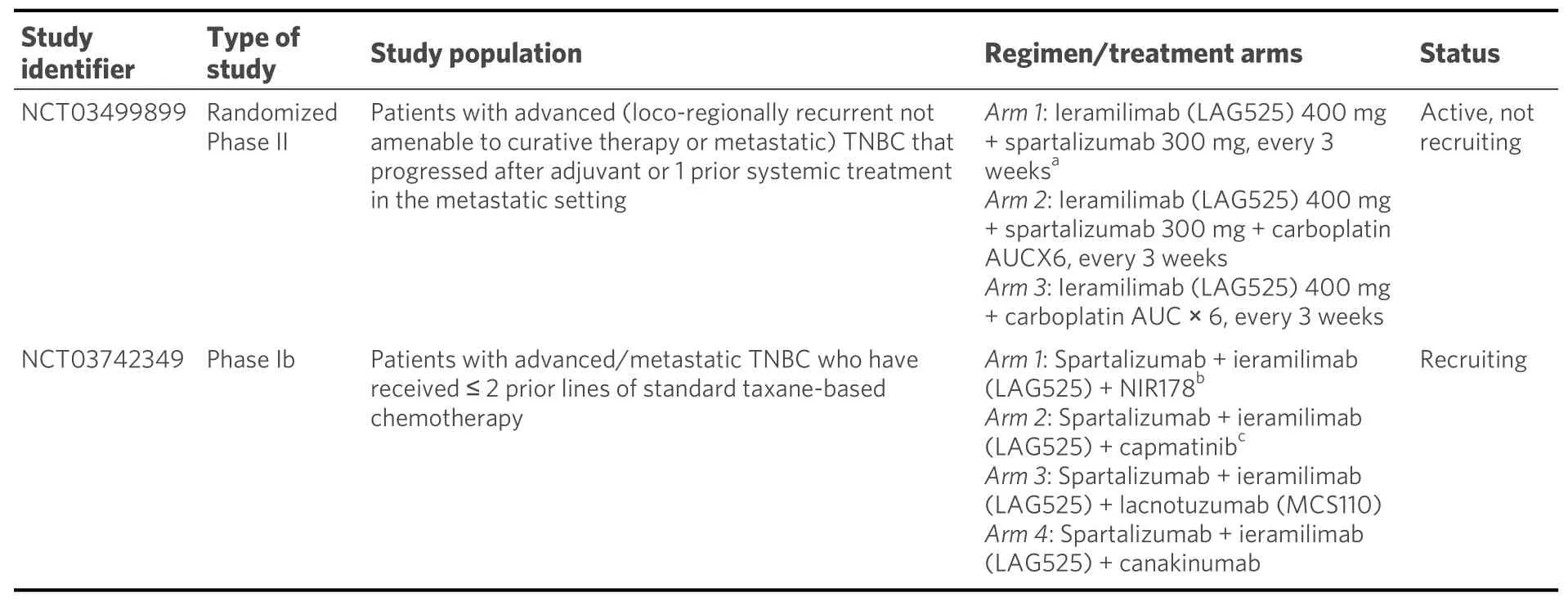
Table 7. Selected current clinical with spartalizumab in combination with other agents in metastatic TNBC
Monoclonal antibodies targeting PD-L1
Avelumab
Avelumab is a human IgG1 moAb that binds to PD-L1 and prevents its interaction with PD-1[243-245]. It has been shownin vitrothat avelumab may also trigger ADCC against a panel of TNBC cells independently of the blockade of the PD-1/PD-L1 axis, and its combination with immunomodulators such as IL-2 or IL-15 could increase the therapeutic efficacy of this immune checkpoint inhibitor[246]. The indications of avelumab include metastatic Merkel-cell carcinoma, locally advanced or metastatic urothelial carcinoma, and advanced renal cell carcinoma in combination with axitinib as first-line treatment[247-251].
1. Avelumab in early breast cancer: neoadjuvant and adjuvant setting
In a pre-therapy proof of concept window study (IMpALA), patients with newly diagnosed TNBC will be treated with one cycle of avelumab either alone or in combination with aspirin[252]. Patients in both arms will also receive lansoprazole (a proton pump inhibitor). It is expected that the combination of avelumab and aspirin will shift the local tumor inflammatory microenvironment towards higher expression of markers associated with effector cytotoxic immunity and clinical benefit. This rationale is based on the knowledge that the production of the inflammatory lipid prostaglandin E2, via cyclooxygenase-2 activity of tumor cells,enables tumor growth through immune escape in pre-clinical models, including TNBC[253-255].
In the adjuvant setting, the very interesting, investigator-driven, phase III A-BRAVE trial[256]randomizes patients who completed treatment with radical intent for primary TNBC, including surgery and chemotherapy, to one year of treatment with avelumabvs. observation. The study enrolls patients in two strata. Patients are stratified to Stratum A if they have completed surgery followed by adjuvant chemotherapy and belong to the following stage categories: pT > 5 cm and pN0, pT > 2 cm and pN1, and any pT with pN2. Patients are stratified to Stratum B if they have completed NACT followed by surgery and did not achieve pCR. Patients who also received additional adjuvant chemotherapy for no more than six months are eligible in Stratum B, after the completion of the adjuvant chemotherapy.
2. Avelumab in mTNBC: monotherapy and combinations with PARP inhibitors
In a phase Ib trial (JAVELIN)[55], patients with metastatic breast cancer, including TNBC, refractory to or progressing after standard-of-care therapy were treated with single-agent avelumab. ORR was 3.0% in the entire population and 5.2% in patients with mTNBC. A trend toward a higher ORR was seen in patients with PD-L1-positivevs. PD-L1-negative tumor-associated immune cells (16.7%vs. 1.6% in the overall population and 22.2%vs. 2.6% in the TNBC subgroup). Treatment-related AEs of Grade ≥ 3 were observed in 13.7% of patients, including two treatment-related deaths.
In another multicohort phase Ib/II trial (JAVELIN PARP Medley)[257], avelumab was combined with the PARPi talazoparib. In the phase Ib study (Cohort 1), patients with advanced solid tumors who had received≥ 1 prior standard-of-care chemotherapy regimens were treated with avelumab in combination with oral talazoparib. The phase II study enrolled patients with either advanced/metastatic TNBC (Cohort 2A) or advanced/metastatic HR+, HER2-, DNA damage repair defect-positive BC (Cohort 2B). Patients in Cohort 2A had received 0-2 prior chemotherapy regimens without progression on prior platinum-based treatment and patients in Cohort 2B had received prior standard of care hormone therapy in either the adjuvant and/or metastatic setting followed by 0-2 prior chemotherapy regimens without progression on previous platinum-based treatment. Twelve patients with advanced cancers were treated in Cohort 1, including two patients with TNBC. Both patients with TNBC had a best overall response (BOR) of SD and remained on treatment for ≥ 9 months. In Cohort 2A, 12 patients were evaluable for disease assessment; BOR was PR in one, SD in six, and PD in five. Adverse events of any grade occurred in 11 patients (91.7%) in Cohort 1 and 18 (94.7%) patients in Cohort 2A. In Cohort 2A, the most common AEs were anemia (57.9%), nausea(26.3%), fatigue (21.1%), and thrombocytopenia (21.1%).
Selected current clinical trials with avelumab in combination with other agents are presented in Table 8.
Atezolizumab
Atezolizumab (MPDL3280A) is a humanized IgG1 moAb that specifically binds to PD-L1[258,259]but does not affect the interaction of PD-1 with its alternative ligand PD-L2, which plays a key role in maintaining immune tolerance[260].endpoints are EFS and pCR (ypT0/Tis ypN0).

?
In the neoadjuvant setting, the phase III NeoTRIPaPDL1/Michelangelo trial[263]randomized 280 patients with TNBC to neoadjuvant carboplatin and nab-paclitaxel with or without atezolizumab. Neoadjuvant treatment was followed by surgery and then four cycles of an anthracycline-based adjuvant regimen. In contrast to KEYNOTE-522, the NeoTRIPaPDL1/Michelangelo trial did not report a favorable outcome with a neoadjuvant immune checkpoint inhibitor with respect to pCR. More specifically, the addition of atezolizumab to NACT for approximately six months resulted in slightly higher rates of pCR when compared to NACT alone in the intent-to-treat population (43.5%vs. 40.8%); however, the increase was not statistically significant. Among patients with PD-L1-positive tumors, pCR rates were 51.9% in the atezolizumab plus chemotherapy armvs. 48.0% in the NACT only arm, but this difference was also not significant.
The findings of the very recently published IMpassion031 trial[264]are of particular interest because they deviate from those of the previously mentioned NeoTRIPaPDL1/Michelangelo trial. The double-blind,phase III, IMpassion031 trial randomized 455 patients with stage II/III TNBC to receive chemotherapy plus atezolizumab or placebo. More specifically, chemotherapy comprised 12 weekly doses of nab-paclitaxel followed by four cycles of doxorubicin plus cyclophosphamide. After the completion of neoadjuvant treatment, surgery followed. Among 333 eligible patients, pCR was documented in 95 (58%) patients in the atezolizumab plus chemotherapy group and 69 (41%) patients in the placebo plus chemotherapy group(difference rate 17%, 95%CI: 6-27; one-sidedP= 0.0044). In the PD-L1-positive population, pCR was observed in 53 (69%) patients in the atezolizumab plus chemotherapy groupvs. 37 (49%) patients in the placebo plus chemotherapy group (difference rate 20%, 95%CI: 4-35; one-sidedP= 0.021). AEs of Grade 3 or 4 were balanced between the two groups.
Finally, a large phase III trial randomizes patients with TNBC to NACT in combination with either atezolizumab or placebo. They will receive 12 doses of weekly paclitaxel plus carboplatin every three weeks,followed by four cycles of AC/EC every two or three weeks. Atezolizumab or placebo will be administered every three weeks during both phases of NACT[265].
In the adjuvant setting, the phase III ALEXANDRA/IMpassion030 trial[266]randomizes patients with stage II or III TNBC to chemotherapy plus atezolizumab or chemotherapy alone. Adjuvant chemotherapy will consist of 12 doses of weekly paclitaxel followed by four cycles of a biweekly anthracycline plus cyclophosphamide regimen. Atezolizumab at 840 mg, every two weeks, will be given concomitantly with chemotherapy, and then at 1200 mg, every three weeks, until completion of one year of atezolizumab.
2. Atezolizumab in mTNBC
A. Atezolizumab monotherapy
A phase I trial (NCT01375842) evaluated atezolizumab in advanced treatment-refractory cancers[267].Among 116 evaluable patients with TNBC, ORR was 24% in first line and 6% in second line or greater.Median duration of response was 21 months, while median PFS was 1.4 months by RECIST criteria and 1.9 months by iRECIST criteria. In first-line patients, median OS was 17.6 months. Patients with PD-L1 expression in ≥ 1% of tumor-infiltrating immune cells had higher ORR and longer OS than those with < 1%expression of PD-L1 (12% and 10.1 monthsvs. 0% and 6.0 months, respectively). Adverse events occurred in 63% of patients and were of Grade 1 or 2 in the majority of them.
B. Atezolizumab with chemotherapy
In a phase Ib trial, women with advanced/metastatic TNBC, who had received ≤ 2 prior lines of chemotherapy in the metastatic setting, were treated with atezolizumab plus weekly nab-paclitaxel. ORR was 39.4% and DCR was 51.5%. Median duration of response was 9.1 months. Median PFS and OS were 5.5 and 14.7 months, respectively. Concurrent nab-paclitaxel neither significantly changed biomarkers of the TME nor impaired atezolizumab systemic immune activation[268].
The phase III IMpassion130 trial[269]evaluated the combination of nab-paclitaxel with atezolizumab as firstline treatment in patients with mTNBC. In total, 902 patients were randomly allocated in a 1:1 ratio to either nab-paclitaxel 100 mg/m2, on Days 1, 8, and 15, plus atezolizumab 840 mg, on Days 1, and 15, every four weeks, or corresponding placebo until disease progression or unacceptable treatment-related toxicities.Primary objectives were PFS and OS, for both the total patient population and PD-L1-positive patients(cutoff ≥ 1%). In the ITT population, the investigator-assessed ORR was 56.0% in the intervention arm, as compared with 45.9% in the control arm (P= 0.002). Furthermore, 7.1% of the patients in the intervention arm had a CR, as compared with 1.6% in the control arm, respectively. In the PD-L1-positive subpopulation, ORR was 58.9% with atezolizumab plus nab-paclitaxelvs. 42.6% with placebo plus nabpaclitaxel (P= 0.002), while the CR rate was 10.3% and only 1.1% in the intervention arm and control arm,respectively (P= 0.002).
At the first interim analysis[269], in the ITT population, median PFS was 7.2 months with atezolizumab plus nab-paclitaxel, as compared with 5.5 months with placebo plus nab-paclitaxel (HR = 0.80; 95%CI: 0.69-0.92;P= 0.002). In the PD-L1-positive subpopulation, median PFS was 7.5 and 5.0 months for patients randomized to atezolizumab and placebo, respectively (HR = 0.62; 95%CI: 0.49-0.78;P< 0.001). At the second interim analysis[270], in the ITT population, median OS was 21.0 months with atezolizumab plus nabpaclitaxel, as compared with 18.7 months with placebo plus nab-paclitaxel (HR = 0.86; 95%CI: 0.72-1.02;P=0.078). In the PD-L1-positive subpopulation, median OS was 25.0 and 18.0 months for patients randomized to atezolizumab and placebo, respectively (HR = 0.62; 95%CI: 0.49-0.78;P< 0.001).
The most common AEs of Grade 3 or 4 included neutropenia in 8% of 453 patients in the atezolizumab armvs. 8% of 437 patients in the placebo arm; peripheral neuropathy (6%vs. 13%); decreased neutrophil count(5%vs. 4%); and fatigue (4%vs. 3%). Treatment-related deaths occurred in two (< 1%) patients in the atezolizumab arm (autoimmune hepatitis related to atezolizumab and septic shock related to nab-paclitaxel)and one (< 1%) patient in the placebo arm (hepatic failure). The data of IMpassion130 confirm the results of the previously mentioned phase Ib trial conducted by Emenset al.[56]. Essentially, the results of IMpassion130 provide evidence of the efficacy of atezolizumab in combination with nab-paclitaxel in patients with PD-L1-positive mTNBC. Therefore, patients’ PD-L1 expression status on tumor-infiltrating immune cells must be taken into consideration when examining treatment options in this subtype of breast cancer characterized by particular therapeutic difficulties in the metastatic stage.
The subgroup analysis of Japanese patients who participated in the IMpassion130 study showed that the efficacy of atezolizumab plus nab-paclitaxel was consistent with that observed in the entire study population. In addition, safety results in the subgroup were consistent with those in the overall population,while Japanese patients had a lower incidence of AEs leading to treatment withdrawal than the overall population[271]. In the context of IMpassion130, Adamset al.[272]published data on patient-reported outcomes. The combination did not compromise patients’ daily functioning or health-related quality of life or worsening treatment symptoms. Finally, a cost-effectiveness analysis of the first-line treatment with atezolizumab plus nab-paclitaxel for advanced/metastatic TNBC showed that the combination is not a costeffective choice in the United States and China[273].
In contrast to IMpassion130, the phase III IMpassion131 trial[274], which compared the efficacy and safety of first-line atezolizumab plus paclitaxelvs. placebo plus paclitaxel in patients with advanced/metastatic TNBC, did not meet statistical significance on its primary endpoint of PFS[275].
Finally, the phase III IMpassion132 trial[276]is of interest, which compares atezolizumab plus chemotherapyvs. placebo plus chemotherapy in patients with progressive disease ≤ 12 months after completing chemotherapy for early TNBC. Investigators can choose as chemotherapy regimen either gemcitabine plus carboplatin or oral capecitabine. Stratification factors are visceral metastases, tumor PD-L1 status, and selected chemotherapy.
Durvalumab
Durvalumab (MEDI4736) is a fully human high-affinity IgG1κ moAb that binds to PD-L1[277,278].Durvalumab was granted accelerated approval by the FDA in May 2017 for the treatment of selected patients with locally advanced or metastatic urothelial carcinoma[279]. Durvalumab was approved by EMA for the treatment of patients with locally advanced, unresectable NSCLC, only if PD-L1 is expressed in ≥ 1%of tumor cells and there was no observable disease progression following platinum-based chemoradiation therapy[280]. Durvalumab has recently been approved by the FDA for use in combination with etoposide and a platinum analog as first-line treatment for patients with extensive-stage SCLC[281].
1. Durvalumab in the neoadjuvant setting: combinations with chemotherapy
A phase I/II trial evaluated the safety and efficacy of concurrent durvalumab with weekly nab-paclitaxel followed by four cycles of dose dense doxorubicin plus cyclophosphamide as neoadjuvant therapy for TNBC. The phase I part of the trial assessed two dose levels of durvalumab. No dose limiting toxicities occurred during this phase I part, and the final overall pCR rate was 44%. In the PD-L1-positive and PD-L1-negative groups, the pCR rates were 55% and 21%, respectively (P= 0.03)[282].
In the neoadjuvant setting, the GeparNuevo phase II trial[283]randomized patients with TNBC to durvalumab or placebo, administered every four weeks, with concomitant weekly nab-paclitaxel followed by four cycles of biweekly epirubicin plus cyclophosphamide. In the window phase, one dose of durvalumab or placebo alone was given two weeks before nab-paclitaxel was started. The pCR rates in the durvalumab and placebo arm were 53.4% and 44.2%, respectively. Of note, durvalumab effect was observed only in the window cohort (pCR 61.0%vs. 41.4%,P= 0.03). In both arms, statistically significantly higher pCR rates were observed in tumors with higher stromal TILs. Furthermore, there was a trend for increased response in PD-L1-positive tumors, which was statistically significant for PD-L1-positive tumor cells in the durvalumab arm (P= 0.045) and for PD-L1-positive immune cells in the placebo arm (P= 0.040). The most common immune-related AE of any grade was thyroid dysfunction.
The phase II PANDoRA trial[284]will randomize patients with TNBC into two treatment arms. Patients in both arms will receive two doses of durvalumab followed by a biopsy, and then they will receive durvalumab with weekly paclitaxel and carboplatin. In Arm B, patients will also receive radiation (24 Gy total) starting with the second durvalumab dose every other day (8 Gy per fraction) for one week. After neoadjuvant treatment, patients will undergo breast surgery and continue on to physician’s choices standard-of-care treatment during the three-year follow-up period.
The open-label, multicenter, phase IIa PHOENIX[285]trial randomizes patients to multiple non-comparative treatment cohorts. The trial consists of two parts: a post-NACT, preoperative window of opportunity component (Part 1) and a postoperative component (Part 2). PHOENIX trial aims to assess whether shortterm exposure to a DNA damage response inhibitor and/or anti-PD-L1 immunotherapy in a preoperative window of opportunity in patients with post-NACT high residual disease load generates a signal of antitumor biological activity within residual TNBC tissue. Patients in Cohort A will receive standard-of-care treatment. Patients in Cohort B will receive AZD6738, preoperatively after NACT in Part 1, and AZD6738,as adjuvant treatment for 12 months in Part 2. AZD6738 (ceralasertib) is an orally available ATR kinase inhibitor that blocks the downstream phosphorylation of CHK1. This prevents ATR-mediated signaling and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and induction of tumor cell apoptosis. Furthermore, AZD6738 sensitizes tumor cells to chemotherapy and radiotherapy[286]. Finally, patients in Cohort C of this trial will receive in Part 1 preoperatively olaparib and in Part 2 adjuvant olaparib for 12 months, while in Cohort D they will receive in Part 1 preoperatively durvalumab and in Part 2 durvalumab for 12 months.
Another ongoing phase Ib/II trial (B-IMMUNE)[287]is evaluating in its phase Ib part the safety and tolerability of durvalumab in combination with a dose dense epirubicin plus cyclophosphamide regimen in the neoadjuvant setting (patients with cT1-cT4 any N, M0, luminal B or triple negative breast cancer).
2. Durvalumab in mTNBC
A. Durvalumab monotherapy as maintenance
The SAFIR02 trial[288]enrolled patients with HER2-negative mBC eligible for first- or second-line chemotherapy. After 6-8 cycles of induction chemotherapy, patients who present an actionable genomic alteration were randomized between targeted therapies and continuation of maintenance chemotherapy,and patients without actionable alterations were randomized between durvalumab for two years and maintenance chemotherapy. The substudy SAFIR02-Immuno trial[289]enrolled 199 patients (HR+ disease 53.4%, TNBC 45.5%) and was powered to show an increment in median PFS of durvalumab over chemotherapy. Median PFS with durvalumab was 2.7 monthsvs. 4.6 months with chemotherapy, at the expense of an increase in the incidence of serious AEs. No subgroup seemed to benefit from immunotherapy. However, median OS was 21.7 months with durvalumabvs. 17.9 months with chemotherapy. This numerical superiority should be attributed to the TNBC and PD-L1-positive subpopulations that appeared to benefit more from durvalumab in the exploratory subgroup analyses.Furthermore, in patients with TNBC, the HR for durvalumab efficacy (OS) was 0.18 for those with CD274 gain/amplification.
B. Durvalumab with other agents
a. Durvalumab with chemotherapy
A phase I/II clinical trial is testing the safety and efficacy of durvalumab in combination with paclitaxel.Thus far, among evaluable patients, the ORR was 42% with a median duration of 7.1 months and DCR of 67%[290].
b. Durvalumab in combination with other immune checkpoint inhibitors
The combination of durvalumab and tremelimumab is described above in the “Tremelimumab” Section[85].
c. Durvalumab with molecular-targeted agents
A phase I study tested in a 3 + 3 dose escalation the combination of cediranib with durvalumab and olaparib[291]. Cediranib is an oral VEGFR1-3 kinases inhibitor[292]. Angiogenesis pathways interact with both DNA repair mechanisms and immune activity. Tumor hypoxia causes downregulation of genes involved in DNA repair, e.g.,RAD51andBRCA1, leading to further DNA damage, genomic instability, and cell death[293,294]. Thus, combining inhibition of DNA repair and angiogenesis pathways may modulate the immune response by increasing DNA damage and tumor mutational burden and attenuating immunosuppressive microenvironments. In this above-mentioned phase I study, which enrolled patients with various malignancies, including TNBC, the recommended phase II dose was tolerable and had preliminary antitumor activity. PD-L1 expression on tumor cells correlated with clinical benefit, but cytokines and peripheral immune subsets did not.
In the phase I/II basket trial MEDIOLA[295], which evaluated the combination of durvalumab with olaparib,patients with various metastatic solid tumors were enrolled into four initial cohorts: germlineBRCAmutated, metastatic breast cancer; germlineBRCA-mutated, metastatic ovarian cancer; metastatic gastric cancer; and relapsed SCLC. Patients with HER2-negative mBC who had received ≤ 2 previous lines of chemotherapy in the metastatic setting were treated with oral olaparib for four weeks, and thereafter with the combination of olaparib plus durvalumab. Among 34 patients enrolled in the study, 16 (47%) had HRpositive breast cancer and 18 (53%) TNBC. Objective responses at Week 12 were observed in 19 (63%)patients including one (3%) complete response. DCR at Weeks 12 and 28 was 80% and 50%, respectively.
CONCLUSION
In the present article, we review major issues related to the development and current use of ICIs in TNBC.Furthermore, we summarize the perspectives on the use of these agents by citing data from active/recruiting trials. Although immunotherapy with ICIs has shown promising results in a variety of malignancies, in mTNBC, i.e., the subtype of breast cancer with the worst outcome, the response rates tο single-agent immune checkpoint inhibitors are very modest, especially when administered to unselected patients. Recent research in TNBC, which is recognized as the most immunogenic among the other breast cancer subtypes,has focused on discovering predictive biomarkers to optimize the use of ICIs, strategies that will make the tumor microenvironment more susceptible to anti-PD(L)-1 treatment, as well other immunotherapeutic strategies beyond ICIs. Clinical trials have established the role of PD-1/PD-L1 inhibition in the first-line treatment in combination with chemotherapy, and atezolizumab plus nab-paclitaxel is the first FDAapproved immunotherapy regimen for breast cancer. In addition to chemotherapy, the combination of ICIs with molecular-targeted agents, including PARP and MEK inhibitors, or with other immunotherapeutic approaches, such as targeting new immune checkpoint targets (LAG-3, TIGIT, VISTA,etc.), cancer vaccines, or immunomodulating agents, appears to have the potential to increase their clinical benefit in TNBC. In addition, in the context of immunogenic cell death, of particular interest is the combination of ICIs with local therapies (radiotherapy and cryotherapy) in both neoadjuvant and metastatic settings.
DECLARATIONS
Authors’ contributions
The collection of data for this review, the writing of the manuscript and its final approval: Papadimitriou M,
Liakouli Z, Papadimitriou CA
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Papadimitriou CA has received speaker honoraria and honoraria for consultancy in advisory boards from Novartis, AstraZeneca, Genesis, MSD, Amgen, Merck and Roche and research grants from BMS and Roche.
The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.
杂志排行

Journal of Cancer Metastasis and Treatment的其它文章
- Pancreatic cancer: genetics, disease progression,therapeutic resistance and treatment strategies
- The unclear role of VEGF in POEMS syndrome:therapeutic implications of neoangiogenesis in a rare plasma cell disorder
- Regulation and function of angiogenic factors in chronic lymphocytic leukemia
- Cancer field surgery for locoregional tumor control of cervical carcinoma
- Uterine cervical carcinoma treated with chemoradiotherapy: impact of three-month MRI follow-up on clinical management and outcome
- AUTHOR INSTRUCTIONS