Regulation and function of angiogenic factors in chronic lymphocytic leukemia
2021-05-11AngelesGarcPardoJavierRedondoMuoz
Angeles García-Pardo, Javier Redondo-Muñoz
Department of Molecular Biomedicine, Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas, Madrid 280140, Spain.
Abstract Progression of chronic lymphocytic leukemia (CLL) is determined by the localization of malignant cells in lymphoid tissues, where they receive growth and survival signals. CLL cells produce angiogenic factors that are regulated by internal and external stimuli and whose levels vary according to the clinical stage of the disease. Stromal cellular and molecular components in CLL niches disturb the balance of pro- and antiangiogenic molecules in CLL cells and induce an angiogenic switch. Additionally, CLL cells also influence the behavior of microenvironmental cells,inducing endothelial cell proliferation and increasing the angiogenic capacity of macrophages, neutrophils, and other cells present in CLL niches. As a result of these reciprocal functional interactions, bone marrow angiogenesis is frequently increased in CLL and has been proposed as a prognostic marker in early disease. Besides their role in regulating angiogenesis, angiogenic factors are also involved in CLL cell migration and survival, all contributing to disease progression. Angiogenic factors, particularly vascular endothelial growth factor, have therefore been attractive therapeutic targets in CLL and many clinical trials were established in the past years. However, the results of these trials reveal that anti-angiogenic therapies alone are not as efficient as expected and should rather be used in combination with other treatments.
Keywords: Chronic lymphocytic leukemia, angiogenic factors, CLL microenvironment, CLL migration and survival,antiangiogenic therapy
INTRODUCTION
Chronic lymphocytic leukemia (CLL), the most common leukemia in Western countries, is characterized by the clonal proliferation and accumulation of CD5+B lymphocytes in the peripheral blood and lymphoid organs[1-4]. The degree of organ infiltration serves to classify CLL into different clinical stages, following the criteria established by Rai (stages 0-IV)[5]and Binet (stages A-C)[6]. According to these criteria, the CLL stage 0/A represents low-risk, I-II/B is an intermediate-risk, and III-IV/C represents high-risk. CLL staging is also useful to determine the need for treatment. Current therapies for CLL include the combination fludarabinecyclophosphamide-rituximab, as well as inhibitors of the B-cell receptor (BCR) signaling pathway, such as ibrutinib/acalabrutinib (Bruton’s tyrosine kinase inhibitors) and idelalisib (phosphatidylinositol 3-kinase-δ inhibitor)[7,8]. An increasing number of CLL cases are now been treated with the Bcl-2 antagonist venetoclax,either as monotherapy or combined with other therapeutic agents[7,8]. Although many patients respond to treatment and some achieve remission, CLL remains an incurable disease.
Clinically, CLL is very heterogenous, with good or poor prognosis mostly determined by the presence of specific markers, particularly mutated (M-CLL) or unmutated (U-CLL) immunoglobulin heavy-chain variable region (IGVH)[1-4]. U-CLL originates from B cells that have not experienced the germinal center and represents an aggressive disease, whereas M-CLL originates from germinal center-differentiated B cells and usually represents a mild form of the malignancy[9]. Other established prognostic markers for CLL are CD38 and CD49d (the α subunit of the α4β1 integrin), whose elevated expression on CLL cells (> 30%) is associated with a poor outcome[10,11]. CLL tumors also accumulate multiple gene mutations and/or deletions,such as those affecting the p53 protein, whose loss of function is related to resistance to chemotherapy[4].Whole-genome sequencing analyses have identified genes that are recurrently mutated in CLL, including notch1 (NOTCH1) and myeloid differentiation primary response gene 88 (MYD88)[12].NOTCH1mutations are mainly detected in U-CLL cases, whileMYD88mutations are predominant in M-CLL[12].NOTCH1mutations are also linked to poor CLL prognosis and high CD49d expression[13]. Recurrent mutations have also been found in genes affecting crucial signaling pathways in CLL, such as those induced by the BCR,NF-κB transcription factor, or MAPK-ERK system[4]. Epigenomic changes are also frequent in CLL, with different patterns in U-CLL and M-CLL cases[4].
CLL progression is determined by the infiltration of bone marrow and secondary lymphoid organs by the malignant cells. This process is mostly mediated by the α4β1 integrin and the chemokine/receptor axes CXCL12/CXCR4 and CCL21/CCR7[14,15]. MMP-9, CD44, particularly the CD44v6 variant, and CD38 have also be shown to play roles in CLL cell migration to tissues[15-17]. Localization in lymphoid niches is beneficial for CLL cells as they receive proliferation and survival signals, which contribute to disease development[18].
Angiogenesis, the development of new vessels from pre-existing ones, is another feature associated to CLL progression[19-21]. Increased angiogenesis has been observed in the bone marrow and lymph nodes of CLL patients[22-24]. The microvessel density in the bone marrow positively correlates with the clinical stage of CLL[22]and has also been associated with a dysregulation of angiogenic factors[25]. Additionally, increased bone marrow angiogenesis in CLL has been suggested as a possible prognostic marker to determine the risk of progression in early disease[26,27].
The angiogenic status of CLL tissues is influenced by cells present in the microenvironment, including CLL cells. Indeed, CLL cells are known to establish reciprocal interactions with stromal cellular components[28-30].These interactions affect cellular functions and modify the CLL cell gene expression pattern, resulting in the so-called “angiogenic switch”[31-33]. This review describes the angiogenic factors produced by CLL cells, their regulation by internal or external factors, and their function in angiogenesis and other cellular processes.We also summarize the anti-angiogenic therapies that have been evaluated in CLL and the results obtained.
CLL CELLS PRODUCE PRO- AND ANTIANGIOGENIC FACTORS AND THEIR RECEPTORS
Angiogenic factors produced by CLL cells
Previous excellent reviews provide a detailed characterization of the angiogenic factors present in CLL and their possible role in the disease[20,21,34,35]. The present review expands and updates the reported studies. CLL cells spontaneously synthesize and secrete pro- and antiangiogenic molecules, including basic fibroblast growth factor (bFGF)[21,34-36], vascular endothelial growth factor (VEGF)[36-38], platelet-derived growth factor(PDGF)[39], thrombospondin-1 (TSP-1)[36], angiopoietin-2 (Ang-2)[36], and matrix metalloproteinase-9(MMP-9)[40-42][Figure 1]. The secreted factors are found in the conditioned media of cultured CLL cells as well as in the plasma/serum and urine of CLL patients, and their levels vary during the course of the disease.Several groups have analyzed samples from CLL patients by enzyme-like immunoassays and have demonstrated that elevated levels of bFGF in lymphocytes and plasma correlate with advanced stages of the disease (Rai stage III/IV)[43-45]. In another study, bFGF levels in urine were also higher in CLL patients than in controls, but they did not significantly correlate with the clinical stage[22].
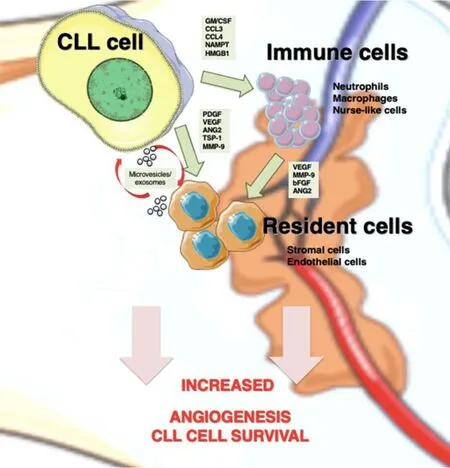
Figure 1. Angiogenesis mediators in CLL niches. In lymphoid tissues, CLL cells establish bidirectional interactions with immune and resident cells. Crosstalk with stromal cells induces an angiogenic switch in CLL cells. Conversely, CLL cells secrete the indicated angiogenic factors, which influence the behavior of immune and resident cells to favor angiogenesis. CLL cells are also able to recruit and induce a proangiogenic phenotype in immune cells by secreting soluble factors, such as GM/CSF (granulocyte-macrophage colony stimulating factor), CCL3 (C-C Motif Chemokine Ligand 3), CCL4 (C-C Motif Chemokine Ligand 4), and NAMPT (Nicotinamide phosphoribosyltransferase). Both CLL and stromal cells also release exosomes, whose cargo (proteins and miRNAs) allows neovascularization and angiogenesis. CLL: Chronic lymphocytic leukemia; PDGF: platelet-derived growth factor; VEGF: vascular endothelial growth factor; ANG2: angiopoietin-2; TSP-1: thrombospondin-1; MMP-9: matrix metalloproteinase-9; bFGF: basic fibroblast growth factor.
VEGF is clearly the most studied angiogenic factor in CLL. The human VEGF family comprises five members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor[46-48]. VEGF-A is the best characterized and comprises five different isoforms (VEGF121, VEGF145, VEGF165, VEGF189, and VEGF206) that arise by alternative splicing of theVEGFgene[46-49]. Using ELISA, Western blot, and RT-PCR analyses of concentrated CLL cell culture media, Chenet al.[38]demonstrated the presence of VEGF121and VEGF165, with molecular weights of 28 and 42 kDa, respectively. VEGF165is the predominant form in CLL and the best studied in terms of expression, regulation and signaling; we refer to this isoform as VEGF throughout this review.
A correlation was found between elevated levels of serum VEGF in early CLL stages (Rai I/II) and the risk of CLL progression/progression-free survival, supporting the role of VEGF as a prognostic marker in this disease[26,50,51]. The amount of plasma PDGF was also higher in CLL patients than in controls and strongly correlated with the levels of VEGF and with advanced stages (Rai II-IV) and poor prognosis markers (ZAP-70 or CD38 positive)[52]. High concentrations of Ang-2 mRNA and plasma protein were also found in several cohorts of CLL patients, with a significant correlation with an aggressive phenotype (unmutated IGHV, CD38 positive), advanced Binet stages (B-C), and shorter survival[53-55]. Similarly, the intracellular and serum concentrations of MMP-9 were higher in CLL than in normal lymphocytes[40-42]. These elevated MMP-9 levels were detectable at early CLL stages[40]and correlated with the risk of disease progression[56,57].In contrast to the above-mentioned factors, the levels of the antiangiogenic molecule TSP-1[58], both mRNA and protein, were higher in low-risk CLL patients (Rai I/II) than in high-risk patients (Rai stage > II)[36]. The same pattern was observed when TSP-1 was quantitated in the conditioned medium of CLL cells cultured for 24 h[36]. These studies indicate that expression and secretion of pro- and antiangiogenic molecules is an active process in CLL, with a clear proangiogenic switch as disease progresses (see below). Quantitation of these factors, however, is not done routinely in the clinic when diagnosing and staging CLL, and their amounts are usually determined as complementary indicators or for specific studies. Accordingly, the levels of angiogenic factors are not commonly included among the criteria used to decide the initiation of treatment in CLL.
Expression of angiogenic factor receptors in CLL cells
bFGF receptors
Besides producing angiogenic/angiostatic molecules, CLL cells also express some of the receptors for these factors. The family of bFGF receptors comprises four members, FGFR1-4, which are present in nearly all hematopoietic cells and mediate multiple physiological processes. In an initial study, Rosenwaldet al.[59]performed gene expression analyses on CLL cells and identified only the FGFR1 transcript, with higher expression in unmutated IgVH cases. However, using Western blotting, immunoprecipitation, and flow cytometry analyses, Sinhaet al.[60]demonstrated that CLL cells expressed elevated levels of FGFR3. Low expression of FGFR1, -2, and -4 was also detected but their levels were similar to those observed on normal B-cells. Additional RT-PCR analyses confirmed the predominant expression of FGFR3[60]. Constitutive phosphorylation at Y653/654 tyrosine residues was also observed in this study, suggesting an active signaling role for FGFR3 in CLL.
VEGF receptors
Initial analyses on a cohort of 216 CLL patients by Western blot showed the consistent expression of VEGFR2 protein, while VEGFR2 was not detected in control samples[61]. Using ribonuclease protection and RT-PCR analyses on CLL cells from 30 patients, Kayet al.[36]demonstrated the presence of mRNA for
VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2). Expression of all three tyrosine kinase VEGF receptors (VEGFR1, VEGFR2, and VEGFR3) at the CLL cell surface was also demonstrated by flow cytometry (13 patient samples) and immunocytochemical (27 patient samples) analyses[62]. The nonenzymatic VEGF receptor neuropilin-1 was also shown to be present in CLL cells, at both mRNA and protein levels, and with a higher expression than in control B-cells[63,64]. VEGF binds to VEGFR1 and VEGFR2 but not to VEGFR3, whose ligands are VEGF-C and -D[46-49]. Although the binding affinity of VEGF to VEGFR1 and VEGFR2 is high, VEGFR2 is the main receptor responsible for delivering intracellular signaling upon interaction with VEGF[20,61]. Therefore, most studies on the CLL system have focused on the functional consequences of the VEGF/VEGFR2 interaction.
The levels of expression of VEGFR2 in CLL cells are variable. Ferrajoliet al.[61]showed that elevated levels of VEGFR2 protein, measured in cell extracts, correlated with shorter survival. An association between high VEGFR2 expression and advanced Rai stages (III/IV) was also observed in that study, although it was not statistically significant. Conversely, we analyzed the constitutive VEGFR2 expression on 38 CLL samples by flow cytometry and did not find a correlation with Rai/Binet stages[65]. Despite this, high expression of VEGFR2 appears to be a characteristic of CLL cells, suggesting an active role of this receptor in the pathology of the disease.
Ang-2 and TSP-1 receptors
The receptor for Ang-2 is Tie-2, one of the two members (Tie-1 and Tie-2) of the Tie receptor tyrosine kinase family[66]. While expression of Tie-1 in CLL cells was early recognized[36,67,68], the expression of Tie-2 on these cells has been controversial. Analyses by PCR and flow cytometry on nine plasma CLL samples failed to show Tie-2 expression, while Tie-2 was present on endothelial cells used as control[69]. Treatment of these CLL cells with various stimuli (CD40L, hypoxia, and stromal/endothelial cells) did not induce Tie-2 expression. In the same study, bone marrow-derived CLL cells expressed Tie-2, detected by PCR and flow cytometry, suggesting a role for the microenvironment in Tie-2 expression on CLL[69].
Aguirre Palmaet al.[68]did not find a constitutive expression of Tie-2 by PCR analyses of peripheral blood CLL cells. However, they observed a transient expression of Tie-2 upon stimulation of these cells with Ang-2. Another study found that a small percentage of peripheral blood CLL cells expressed Tie-2 at the cell surface[70]. Because Tie-2 undergoes rapid internalization upon activation[71]these authors performed intracellular staining and found a moderate positive expression of Tie-2 receptor. CLL cells infiltrating lymph nodes also expressed Tie-2. The presence of Tie-2 was further confirmed by Western blotting and PCR analyses[70]. It therefore appears that expression of the Tie-2 receptor in CLL cells is subjected to modulation by several external factors.
Two main TSP-1 receptors, CD36 and CD47, are expressed by CLL cells. CD36 expression was initially studied by Rutellaet al.[72]on 24 CLL samples. Using flow cytometry and immunofluorescence analyses,they found variable expression of CD36 in most CLL cases. Elevated CD36 expression was associated with advanced disease (Rai stage III/IV, organ infiltration)[72]. CD47 was also shown to be present in CLL cells and to mediate several functions upon binding to TSP-1, including cell death and cytoskeleton reorganization[73,74].
MMP-9 receptors
Although MMP-9 is mainly a secreted protease found in soluble form in serum and cell conditioned media,we and others have consistently found MMP-9 at the CLL cell surface[40-42,75]. Using immunoprecipitation with anti-MMP-9 antibodies, function blocking antibodies, siRNA transfection, and immunofluorescence analyses, our group demonstrated that binding of MMP-9 to CLL cells is mediated by a docking complex consisting of α4β1 integrin and a 190 kDa CD44v isoform[75]. Interestingly, silencing either α4β1 integrin or 190 kDa CD44 with specific siRNAs reduced the amount of cell surface-bound MMP-9 and increased the levels of secreted MMP-9, indicating that there is a dynamic traffic between soluble and cell-bound MMP-9 in CLL. Perhaps this could explain why we could not find in our studies a clear association between the expression of membrane-bound MMP-9 and the clinical stage of the disease[76-79]. The ability to bind and retain MMP-9 at their surface appears to be characteristic of CLL cells as it was not observed in normal Bcells[75].
α4β1 integrin is present in approximately 40% of CLL cases, and there is now extensive evidence showing that the α subunit of this integrin, also known as CD49d, is a strong independent prognostic marker in CLL,associated with an aggressive disease[10,80]. Our studies have shown that, as a receptor for MMP-9, α4β1 integrin contributes to disease progression by regulating CLL cell migration and survival[77-79,81]. This contribution was recently confirmed based on a bimodal pattern of expression of CD49d in CLL cells[82].CD44 (standard and variant forms) is expressed at high levels in CLL cells and has also been proposed as a prognostic marker, particularly due to its role in CLL cell migration and organ localization[17].
REGULATION OF ANGIOGENIC FACTORS IN CLL
Regulation by conditions of the microenvironment - hypoxia
The constitutive production of angiogenic molecules and their receptors by CLL cells is subjected to regulation by several stimuli. Because progression of CLL is characterized by the migration and localization of malignant cells in lymphoid tissues[18,30], these stimuli are mainly provided by the microenvironment of these tissues. Hypoxia, a common condition in human bone marrow and other tissues, is a key regulator of VEGF in physiological and pathological conditions[20,47,49]. Indeed, hypoxia stabilizes the hypoxia-inducible factor-1α (HIF-1α), a major transcription factor for VEGF, allowing its binding to specific elements in the VEGF promoter and increasing VEGF synthesis and secretion[47,49]. Several authors have shown that, upon a hypoxic stimulus, cultured CLL cells increase their VEGF production, at both mRNA and protein levels[36,38]. Moreover, we and others demonstrated by immunohistochemical analyses that VEGF is present in the bone marrow and lymph nodes of CLL patients, and at higher expression than in normal tissues[38,65].Kayet al.[36]found that culturing CLL cells under hypoxic conditions also reduced the levels of TSP-1 produced by these cells, thus favoring an angiogenic switch. The levels of PDGF and Ang-2 in CLL cells were also shown to be upregulated by hypoxia[36,52,83].
Regulation by stromal cells - induction of a proangiogenic phenotype on CLL cells
Upon migration and localization in lymphoid tissues, CLL cells interact with surrounding stromal cells,establishing an active crosstalk that provides survival and proliferation signals to the malignant cells. The molecular bases of these interactions have been recapitulated in excellent previous reviews[30,84]and involve direct cell-cell contact via adhesion molecules, release of soluble factors such as chemokines, or material exchange via extracellular vesicles [Figure 1]. One important consequence of these cellular interactions is the modification of the gene expression profile of CLL cells, mainly resulting in the upregulation of antiapoptotic molecules (Bcl-2, Bcl-xL, Mcl-1, and XIAP) and the activation of survival signaling pathways(PI3-K/NF-κB and Notch)[30]. Additionally, the balance of pro- and antiangiogenic factors on CLL cells is also affected by the contact with stromal cells. Kayet al.[31]showed that culturing CLL cells on primary bone marrow-derived stromal cells dramatically increased the secretion of bFGF and reduced the levels of the antiangiogenic molecule TSP-1. In another study, Edelmannet al.[32]performed gene microarray analyses on CLL cells that had been co-cultured with the murine bone marrow fibroblast cell line M2-10B4, either in direct cell-cell contact or separated by Transwell inserts. They found that direct contact with the fibroblastic cells significantly increased VEGF expression at both gene and protein levels and decreased TSP-1 expression. Consistent with this decrease, there was a correlation between advanced CLL (Binet stages B and C) and low TSP-1 plasma levels, in agreement with the studies mentioned above. Additionally, MMP-9 was also dramatically upregulated (25.9-fold change) upon CLL cell contact with the fibroblastic cells, compared with freshly isolated CLL cells[32], a fact also confirmed by Schulzet al.[85]. In line with this upregulation, we showed that culturing CLL cells on primary stromal cells (derived from a CLL patient) for 48 h increases the amount of MMP-9 bound to the CLL cell surface by two-fold[81]. Moreover, CLL cells isolated from the bone marrow or lymph nodes of patients consistently showed higher levels of surface-bound MMP-9 than their peripheral blood counterparts, confirming the increased production of MMP-9 in a pathophysiological context[81]. This MMP-9, which is produced by the CLL cells as well as by stromal cells, likely contributes to the increased angiogenic status observed in CLL tissues (see below).
Endothelial cells are also an important stromal component of lymphoid tissues [Figure 1]. Maffeiet al.[33]showed that physical interaction of CLL cells with human umbilical vein endothelial cells protected CLL cells from spontaneous and drug-induced apoptosis. This interaction was mediated by β1 and β2 integrins,and it also resulted in the modulation of 1944 genes, 1217 upregulated and 727 downregulated, determined by gene microarray analyses[33]. Many of the upregulated genes were related to angiogenesis and included the Ang-2 receptors Tie-1 and Tie-2, VEGFC, TSP-1, MMP-2, and MMP-14, while VEGFR3 was downregulated. The C-C motif chemokine-2 (CCL2), which is known to regulate angiogenesis and recruit monocytes/macrophages to tissues[86], was also significantly upregulated in CLL cells upon coculture with endothelial cells[33]. It can be concluded that, in lymphoid tissues, particularly in the bone marrow, these CLL-produced factors disturb the angiogenic balance and contribute to disease progression.
Autocrine and inter-regulation among angiogenic factors
The fact that CLL cells express angiogenic factors and their receptors supports the existence of autocrine and crosstalk regulations of the expression and function of these factors. These regulations may take place on circulating CLL cells as well as in CLL cells located in niches. On isolated CLL cells, Bauvoiset al.[40]showed that antibodies to VEGF decreased MMP-9 expression, suggesting a link between both proteins. We showed that binding of VEGF to its VEGFR2 receptor in CLL cells downregulates the expression of MMP-9 as well as the migration of these cells through Matrigel or endothelial cells[87]. Regulation of MMP-9 by the VEGF/VEGFR2 axis was at the transcriptional level and was mediated by the phosphorylation and translocation to the nucleus of the signal transducer and activator of transcription 1 (STAT1)[87], a factor known to suppressMMP-9gene transcription in response to interferons in several cell systems[88].
Using immunofluorescence analyses, our group also demonstrated that MMP-9 is present in the bone marrow and lymph nodes of CLL patients, in partial association with the macrophages in these tissues[79].We studied whether, as a component of the CLL microenvironment, MMP-9 affected the CLL cell angiogenic pattern. Culturing CLL cells on immobilized MMP-9 for 24 h increased the expression of MMP-9 and VEGF and reduced the expression of the angiostatic molecules TSP-1 and Ang-2, all at the gene and protein levels[79], establishing that isolated MMP-9 is able to induce a proangiogenic profile in CLL cells. The fact that MMP-9 interaction with its receptors in CLL cells increases its own production may represent an autocrine positive feedback loop, similar to that described for VEGF in these cells[20].
Further mechanistic studies demonstrated that downregulation of TSP-1 by MMP-9 involved α4β1 integrin(MMP-9 receptor), Src kinase family activity, and the STAT3 transcription factor[79]. The fact that STAT3 is also a transcription factor for VEGF, regulates the expression and degradation of HIF-1α, and is a key factor in angiogenesis[89-92]strongly suggests that upregulation of VEGF by MMP-9 is also mediated by STAT3 activation.
Other levels of regulation - epigenetic and gene polymorphism
Angiogenic molecules in CLL cells can also be regulated through other molecular mechanisms, including gene polymorphisms and epigenetic alterations. The genomic polymorphism in VEGF-A of CLL cells has been extensively studied. According to Lozano-Santoset al.[93], a specific VEGF haplotype correlates with shorter survival in CLL patients. Furthermore, patients who carry this haplotype in combination with other clinical features, such as negative CD38 expression or early age at diagnosis, show a poor survival ratio[93].Other studies have also indicated that VEGF polymorphism might be relevant as a genetic risk and adverse survival marker in CLL[94,95]. On the other hand, epigenetic profiles might serve to stratify patients and identify different molecular pathways involved in CLL progression. For example, the expression of theAng-2gene promoter in CLL cells is highly dependent on the DNA methylation status[96]. This methylation is regulated by VEGF, which promotes Ang-2 expression and neovascularization[96]. In agreement with the notion that DNA methylation regulates critical steps during CLL angiogenesis, it has been reported that Ang-2 expression is also regulated by the tumor suppressor gene microcephalin[97]. Furthermore, other angiogenic molecules such as endothelin-1 are also regulated by DNA methylation in CLL cells[98].
CONTRIBUTION OF CLL CELLS TO THE ANGIOGENIC STATUS OF LYMPHOID TISSUES
Functional effects on endothelial cells
The above-mentioned studies have clearly established that the molecular and cellular components of the microenvironment in CLL tissues modulate the gene expression pattern of CLL cells, inducing a prosurvival and proangiogenic phenotype in the malignant cells. It is also known that modulation of angiogenic molecules in CLL has functional consequences in the CLL niches [Figure 1]. Chenet al.[38]showed that the conditioned medium (CM) of CLL cells cultured for 24 h contained VEGF and induced proliferation of endothelial cells. Moreover, this CM also induced moderatein vitroangiogenesis, and this effect was prevented by a neutralizing anti-VEGF antibody[38]. It was later demonstrated that the CM of cultured CLL cells also contained Ang-2, and the levels of this protein correlated with the degree of bone marrow vascularization of the patients studied[84]. These authors also showed that both Ang-2 and VEGF were responsible for the CM-induced tube formation by endothelial cells on Matrigel matrices, as neutralizing antibodies to either factor diminished the angiogenic effect[83]. In both studies, a hypoxic stimulus further increased the functional effects of the CLL cell CM on endothelial cells. Additionally, we showed that the CM of CLL cells that had been incubated with MMP-9 for 24 h had a significantly higher effect on endothelial cell proliferation compared to the CM of CLL cells incubated on 0.5% BSA[79]. This is likely due to the MMP-9-induced production of proangiogenic factors (MMP-9 and VEGF) by CLL cells mentioned above.
Functional effects on mesenchymal stromal cells
Besides affecting endothelial cell behavior, CLL cells also influence the function of other cells present in the microenvironment of CLL niches[29]. An active crosstalk between CLL and mesenchymal stromal cells(MSC), resulting in malignant cell survival and changes in the pattern of cytokine production by MSC, has been reported[99]. Moreover, the CM of CLL cells was shown to drive migration and proliferation of MSC[52].This effect was mediated by the interaction of the PDGF present in the CM of CLL cells with its PDGFR in mesenchymal cells, which resulted in activation of the receptor and induction of Akt phosphorylation.Binding of the CLL-derived PDGF to PDFGR also induced the production of VEGF (but not bFGF or TSP-1) by mesenchymal cells through a PI3-K-dependent mechanism[52]. This is a clear example of how CLL cells actively modulate their microenvironment, inducing an angiogenic switch that is permissive for CLL progression.
Functional effects on macrophages
Macrophages are also important components of tissue microenvironments. In CLL, the specific population of leukemia-associated macrophages, known as nurse-like cells, plays a fundamental role in inducing and maintaining CLL cell proliferation and survival[29,100,101]. The important function of tumor-associated macrophages in angiogenesis is also well documented[102,103]. Macrophages can be activatedin vitroby specific stimuli towards two different functional phenotypes: M1 and M2[104]. M1 macrophages are proinflammatory and induce strong antitumor immune responses by producing relevant specific molecules and cytotoxic factors. In contrast, M2 macrophages are anti-inflammatory and secrete chemokines/cytokines that suppress immune responses and support tumor expansion[105,106].In vivoevidence indicates that tumorassociated macrophages are mostly polarized towards the M2-like phenotype and that tumor cells contribute to this polarization[107]. In the case of CLL, Audritoet al.[108]showed that activated CLL cells induced monocyte differentiation to M2 macrophages, and this was mediated by the nicotinamide phosphoribosyltransferase enzyme produced by CLL cells [Figure 1]. These M2 macrophages sustained CLL cell survival and reduced T-cell proliferation, thus favoring disease progression[108]. In another study, the high-mobility group protein B-1 produced by CLL cells was shown to differentiate nurse-like cells to M2-polarized macrophages, concomitant with a STAT3 and NF-κB activation on both cell types[109]. The hypoxic conditions found in CLL niches also indirectly induced M2 macrophage polarization via increase production of adenosine[29].
Polarization of macrophages towards the M2 subtype also has important consequences for angiogenesis.Zajacet al.[110]showed that M2 macrophages have higher proangiogenic capacity than the M1 subtype, due to the increased production of MMP-9 free of tissue inhibitor of metalloproteinases-1 (TIMP-1), the specific MMP-9 inhibitor. Indeed, these authors showed that M2 macrophages shutdown theirTIMP-1gene expression, a fact that was also demonstrated with murine bone marrow-derived macrophages. The importance of MMP-9 for M2 macrophage angiogenic function was further demonstrated by showing thatMMP-9-null M2 macrophages were not angiogenic, despite their downregulation of theTIMP-1gene[110].Moreover, the same group showed that neutrophils, which do not produce TIMP-1[111], are the major source of proangiogenic MMP-9 in tumor tissues[112]. MMP-9 is present in CLL tissues, where it is produced by the mentioned stromal cellular components as well as by CLL cells. Our group demonstrated that the CM of 24 h CLL cell cultures induced a proangiogenic profile in both M1 and M2 macrophages after 7 days of culture, as determined by the increased expression of MMP-9 in these cells, at both mRNA and protein levels[79]. Although we did not test whether the MMP-9 produced by M2 macrophages was more angiogenic than that produced by M1 macrophages, this study constitutes another example of how CLL cells modulate the angiogenic profile of microenvironmental cells in CLL tissues.
Functional effects on neutrophils
The role of neutrophils in tumor initiation and angiogenesis is well recognized[113]. In solid tumors, such as melanoma, hepatocellular carcinoma, and breast carcinoma, the recruitment of proangiogenic neutrophils triggers STAT3 activation and the production of angiogenic molecules, including VEGF and MMP-9[114].There is also a proangiogenic subpopulation of neutrophils, which express high levels of α4β1 integrin,CXCR4, and MMP-9 and are recruited to hypoxic tumor sites[115]. Indeed, as mentioned above,inflammatory neutrophils are the major producers of angiogenic MMP-9 in tumor tissues[112]. In the case of CLL, neutrophils were shown to be activated, expressing high levels of CD54 and presenting some functional defects[116]. Podazaet al.[117]showed that CLL cells induced neutrophil survival and their reprogramming to an immunosuppressive phenotype. In turn, neutrophils were shown to affect CLL cell activation and survival through the induction of extracellular traps[117], thus establishing a reciprocal cellular interaction that supports the proangiogenic function of neutrophils in CLL niches.
Functional effects of CLL cell-derived exosomes
Exosomes are small extracellular vesicles secreted by normal and malignant cells via exocytosis in response to multiple physiological and pathological conditions[118]. Exosomes contain proteins, DNA, mRNAs and non-coding RNAs (such as miRNAs) that play fundamental roles in cell-cell interactions and tumorinduced microenvironment modifications[119]. Exosomes released by CLL cells were shown to induce the transition of mesenchymal stromal cells to cancer-associated fibroblasts, which support the growth and survival of malignant cells[120]. These exosomes also impacted on endothelial cells increasing angiogenesis in CLL tissues[120]and modulated intracellular signaling pathways on stromal cells, all favoring disease progression[121,122].
Analysis of the proteomic and miRNA profiles from exosomes produced by CLL cells revealed the presence of several cell surface proteins (CD9, CD37, CD53, CD63, and CD82) and miRNAs, including miR-21,miR-146a, miR-29 family, miR-150, miR-155, and miR-223[120,123][Table 1]. The expression of some of these miRNAs increases upon BCR activation and has been associated with CLL pathogenesis[123]. Moreover, the expression levels of miR-155 in plasma exosomes were proposed to be a biomarker for the risk of progression from monoclonal B-cell lymphocytosis to CLL, as well as for the identification of CLL patients who may show resistance to therapy[124]. In another study, Farahaniet al.[125]showed that exosomes released by CLL cells altered the transcriptome of the stromal cell line HS-5, a fact that may influence the prosurvival signals induced by these cells which favor CLL progression[86].
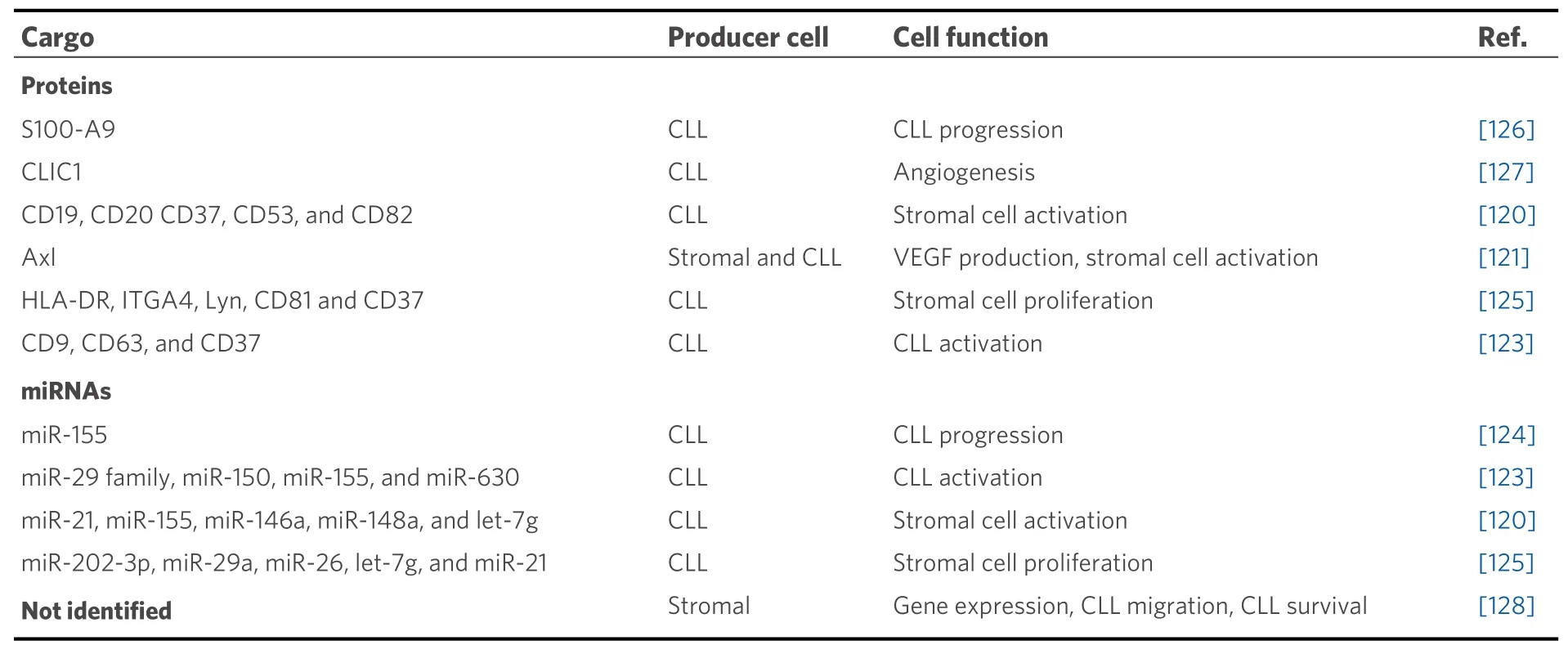
Table 1. Molecular components of stromal and CLL-derived exosomes
CLL cell-derived exosomes also contain proteins that affect the molecular pathways of recipient cells,including those involved in angiogenesis. For instance, they contain the small calcium-binding protein S100-A9, which regulates the PI3-K/Akt/NF-κB signaling pathway, VEGF production, and inflammatory processes[126]. Another protein present in CLL exosomes is Axl, a receptor tyrosine kinase, which induces stromal cell activation, VEGF production, and CLL progression[121]. Moreover, the chloride intracellular channel 1 protein, also found in CLL exosomes, was recently shown to induce endothelial cell proliferation and angiogenesis through upregulation of β1 integrin and the MAPK/ERK signaling pathway[127]. It has also been reported that extracellular vesicles from bone marrow stromal cells modify the gene expression pattern in CLL cells, affecting BCR activation and other processes[128]. Uptake of these vesicles also rescued CLL cells from apoptosis and enhanced their migratory capacity in response to the CXCL12 chemokine[128].Altogether, extracellular vesicles produced by CLL or stromal cells constitute another important way of tumor-microenvironment communication in tissues, with consequences on many processes including angiogenesis in CLL niches. These vesicles thus represent potential therapeutic targets, and several clinical trials are currently addressing this possibility[30].
OTHER FUNCTIONS OF ANGIOGENIC FACTORS IN CLL
Besides contributing to the observed increased angiogenesis in CLL niches, several angiogenic factors produced by CLL cells are also able to perform other functions that directly affect disease progression. The best studied functions are the role of these factors in CLL cell migration and survival.
Function of angiogenic factors in CLL cell migration
VEGF
A role of VEGF in CLL cell migration was first inferred by the fact that blocking autocrine VEGF or the VEGFR2 kinase activity reduced the chemokine-induced CLL cell migration through endothelium[129]. This study also showed that chemokine activation of αLβ2 integrin and subsequent transendothelial migration was defective in CLL cells and that stimulation with autocrine VEGF and α4β1 integrin overcame this defect and restored cell motility. Regulation of cell motility by these two proteins appears to be particularlyrelevant in the migration of CLL cells to lymph nodes, as it was preferentially observed in patients with lymphadenopathy[129]. Another example of VEGF involvement in CLL cell migration is the previously mentioned study[88], in which we showed that binding of exogeneous VEGF to VEGFR2 reduces CLL cell migration in a dose-dependent manner. This effect was mediated by the VEGF/VEGFR2-induced downregulation of MMP-9[88].
MMP-9
It is well established that MMPs facilitate cell migration by degrading basement membranes and extracellular matrices, as well as by releasing matrix-bound growth factors and chemokines[130]. It is also well established that many MMPs may display non-catalytic activities, mostly by localizing at the cell surface,either via transmembrane domains (MT-MMPs) or by binding to specific cell surface receptors[131]. MMP-9 is the main MMP expressed by CLL cells and localizes at the cell surface by binding to the α4β1 integrin/CD44v complex[75]. Our group showed that CLL migrationin vitroas well asin vivohoming to bone marrow and spleen requires optimal MMP-9 expression, and that above these optimal levels migration is inhibited[75,77]. This was demonstrated using the CLL-derived MEC-1 cell line stably transfected with MMP-9 or empty vector as control, as well as primary CLL cells previously incubated with MMP-9[77].Inhibition of CLL cell migration by elevated concentrations of MMP-9 was partly due to the downregulation of migration regulatory pathways such as those involving the GTPase RhoA and the kinases Akt, ERK, and FAK, together with the concomitant upregulation of p190RhoGAP (RhoA inhibitor) and PTEN (Akt/ERK/FAK inhibitor)[77]. Moreover, a proteolytically inactive MMP-9 mutant had a partial migration inhibitory effect, indicating that both catalytic and non-catalytic MMP-9 functions were involved[132]. Notably, the dual regulatory role of MMP-9 likely operatesin vivosince: (1) CLL cell interaction with stroma increases cell-bound MMP-9[32,81,85]; (2) CLL cells from lymphoid tissues express more MMP-9 than their peripheral blood counterparts[81]; and (3) MMP-9 is present in CLL tissues[79].Upregulation of MMP-9 in CLL cells and tissues is also induced by α4β1 integrin ligation, chemokine(CXCL12 and CCL21)-receptor interactions, or CD38 interactions[15,42,133]. Elevated levels of MMP-9 in lymphoid tissues would therefore favor the retention of CLL cells in these niches and contribute to disease progression.
Function of angiogenic factors in CLL cell survival/apoptosis
VEGF
Binding of many angiogenic factors to their respective receptors in CLL cells are known to induce signaling pathways that lead to apoptosis resistance and cell survival[20,21,34,35]. It was first demonstrated that culturing CLL cells with VEGF for 24 h significantly decreased spontaneous and chrorambucil-induced apoptosis,and this involved upregulation of the anti-apoptotic molecules myeloid cell leukemia-1 (Mcl-1) and Xlinked inhibitor of apoptosis protein (XIAP) via activation of the transcription factor STAT3[134,135]. These authors also showed that the VEGFR1 and VEGFR2 receptors are constitutively phosphorylated in CLL cells and that tyrosine kinase inhibitors or anti-VEGF antibodies inhibited this phosphorylation and STAT3 activation, reduced Mcl-1 and XIAP levels, and induced apoptosis[134,135]. These studies substantiate the presence of a VEGF/VEGFR autocrine pathway that supports CLL cell survival and demonstrate that blocking this pathway results in CLL cell apoptosis. Interestingly, it was further demonstrated that this internal autocrine VEGF survival loop particularly operates in CLL cells expressing the prognostic marker CD38, thus introducing a different VEGF behavior in CD38+ and CD38- CLL cells[136].
Autocrine VEGF was also shown to mediate the survival effect of CD40 ligand (CD154), a molecule expressed on activated T cells, monocytes, macrophages, and other components of the microenvironment[137]. These authors demonstrated that the anti-apoptotic effect of CD154 required the cooperative signaling provided by both CD40 and VEGFR1, 2, resulting in NF-κB activation and upregulation of the protein survivin[137]. Functional cooperation of VEGF receptors and other cell surface molecules, such as integrins, has been clearly established in the context of endothelial cells and tumor angiogenesis[138]. In CLL cells, we demonstrated that the α4β1 integrin is associated with VEGFR2 and modulates VEGF functions on these cells, including the survival effect induced by exogenous VEGF[65].
bFGF
In initial studies, it was shown that elevated intracellular levels of bFGF correlated with CLL cell resistance to fludarabinein vitroand that addition of bFGF delayed apoptosis in fludarabine-treated cells[44]. Using CLL-derived cell lines and primary CLL cells, it was also demonstrated that bFGF delayed fludarabineinduced apoptosis by upregulating Bcl-2, at both mRNA and protein levels[139]. Bcl-2 expression was also shown to positively correlate with the levels of bFGF in serum in the 85 patients studied[140]. In another study, Romanovet al.[141]reported that bFGF suppressed p53 activation in cultured CLL cells exposed to ionizing radiation, mainly by upregulating the p53-inhibitory protein MDM2 (mouse double minute 2).bFGF may thus induce CLL cell survival by several mechanisms. Moreover, the survival activity induced by bFGF interaction with its FGFR3 receptor in CLL cells can be potentiated by the tyrosine kinase Axl, which forms a complex with FGFR3 and modulates its signaling pathway[60].
Ang-2 and TSP-1
It was initially reported that Ang-2 induces CLL cell survival after 24 h but that, at longer times, Ang-2 has a pro-apoptotic activity[68]. This was attributed to the transient modulation of the Tie-2 receptor, as well as of caspases and ATP content[68]. However, another study has shown that the Ang-2/Tie-2 axis induces CLL cell survival even after 96 h of treatment[70]. These authors also showed that Tie-2 engagement by Ang-2 activated the PI3-K-Akt signaling pathway, and this was abolished by a Tie-2 kinase inhibitor, which, in fact, induced apoptosis[70]. This study therefore supports an autocrine role of Ang-2 which affects CLL cell survival.
In contrast to the described pro-survival function of some angiogenic factors, binding of TSP-1 to its receptor CD47 was shown to induce apoptosis in CLL cells[73]. The mechanism involved in this action was independent of caspase activation and was not overcome by survival factors such as IL-4 or γ-interferon. It was later demonstrated that induction of apoptosis by TSP-1-CD47 interaction was mediated by cytoskeleton reorganization, mainly involving the small GTP binding protein Cdc42 and the Wiskott-Aldrich Syndrome protein signaling pathway[74]. Another study has shown that peptides derived from the Cterminal domain of TSP-1 targeted CD47 and efficiently induced apoptosis of CLL cells[142]. This apoptotic effect involved activation of phospholipase C γ-1 and was also observedin vivowhen injecting these peptides in a CLL xenograft murine model[142].
MMP-9
A role for MMP-9 in CLL cell survival was first demonstrated in co-cultures of CLL and bone marrow stromal cells[143]. These authors showed that blocking the constitutive MMP-9 activity with neutralizing antibodies suppressed the anti-apoptotic effect of stromal cells. We reported that culturing CLL cells on immobilized or soluble MMP-9 prevented their spontaneous apoptosis[81]. The MMP-9 effect was dosedependent and persisted for the seven days of the assay. Importantly, induction of cell survival did not involve the proteolytic activity of MMP-9 but was mediated by MMP-9 binding to α4β1 integrin and CD44v at the CLL cell surface. Indeed, a catalytically inactive MMP-9 mutant or the isolated hemopexin domain of MMP-9 fully reproduced the survival-supporting function of the molecule. This study further showed that MMP-9 (or its hemopexin domain) binding to these receptors induces an intracellular signaling pathway consisting in Lyn activation, STAT3 phosphorylation, and Mcl-1 upregulation. This pathway was observed in all CLL cases studied and was active in CLL lymphoid tissues[81].
Besides preventing spontaneous apoptosis, MMP-9 also protected CLL cells from the apoptotic effect of cytotoxic drugs, such as fludarabine or arsenic trioxide[76]. This study showed that both of these compounds transcriptionally upregulated MMP-9, which mainly localized at the CLL cell surface. Blocking MMP-9 with antibodies reduced the protective effect of stromal cells to the apoptotic function of arsenic trioxide or fludarabine.
Additionally, MMP-9 induced CLL cell drug resistance by modulating the balance of anti- and proapoptotic proteins of the Bcl-2 family towards an anti-apoptotic pattern[76]. Induction of cell survival therefore represents another contribution of MMP-9 to CLL progression.
Other possible roles of angiogenic factors in CLL
During the course of CLL, approximately 2%-10% of patients develop an aggressive lymphoma, known as Richter’s transformation or Richter’s syndrome[144]. In the CLL phase, risk factors for Richter’s transformation include an advanced clinical stage, presence of poor prognostic markers (unmutated IGVH,CD38, and CD49d), and genetic aberrations/mutations of specific genes (TP53,NOTCH1, andMYC)[144].Other factors, however, may certainly contribute to the CLL-lymphoma transformation. A recent study used patient-derived samples and murine animal models and elegantly demonstrated that activation of Akt triggers Richter transformation via induction of Notch1 signaling[145]. Although the possible involvement of angiogenic factors in Richter’s transformation has not been directly addressed, it is well known that activation of the PI3-K/Akt pathway is a major signaling event upon binding of several angiogenic factors(VEGF, bFGF, PDGF, and Ang-2) to their respective receptors[48,49,52,70]. It is therefore possible that these factors, which, as explained above, are present at elevated levels in advanced CLL, contribute to Richter’s transformation by activating Akt.
ANTIANGIOGENIC THERAPY IN CLL
Despite the development of many new therapies in the last decades targeting specific pathways or molecules, CLL remains an incurable disease. Angiogenesis is a common process in cancer, which helps the growth and dissemination of malignant cells. As in other tumors, angiogenesis provides CLL cells with nutrients and survival signals to facilitate disease progression[20-22]. Targeting angiogenesis thus seems a useful therapeutic strategy against CLL [Table 2]. This section summarizes the results obtained with these antiangiogenic treatments in CLL.

Table 2. Tested reagents in antiangiogenic therapy for CLL
Fludarabine
Fludarabine is a purine analogue and a frontline treatment in CLL, alone or combined with other compounds such as rituximab and/or cyclophosphamide[1,7,8,146]. The antiangiogenic role of fludarabine was first assessed by Molicaet al.[147]who measured the microvessel density in the bone marrow samples of CLL patients and found that this density decreased after fludarabine treatment. The same authors later evaluated the effect of combining fludarabine with alemtuzumab (anti-CD52 antibody) and found an even more significant reduction of angiogenesis in the bone marrow of CLL patients who had received this treatment[148]. In another study, the combination of fludarabine with the γ-secretase inhibitor PF-03084014 resulted in a synergistic induction of apoptosis and the impairment of angiogenesis and cell migration[149].Importantly, these synergistic effects were specific for Notch1-mutated CLL cells, known to represent a high-risk form of the disease[149].
Endothelin receptor inhibitor
CLL cells express high levels of endothelin receptors, which regulate CLL cell-microenvironment interactions and angiogenesis[98]. Maffeiet al.[150]reported that inhibiting endothelin receptors with macitentan impaired CLL cell survival, migration, and proliferation. Macitentan also reduced VEGF expression by decreasing HIF-1α accumulation, thus directly affecting angiogenesis[150].
Lenalidomide
The use of inhibitors of the transcription factor NF-κB for the treatment of hematological tumors has been proposed for a long time. Thalidomide was a first-generation drug which downmodulated the levels of angiogenic molecules, such as tumor necrosis factor-α and VEGF[151,152]. Indeed, these two angiogenic factors were downregulated in CLL patients treated with thalidomide in combination with fludarabine[153].Lenalidomide is a thalidomide analog frequently used as a therapeutic agent in hematological malignancies,including multiple myeloma, myelodysplastic syndrome, and CLL[154]. In CLL, lenalidomide was shown to be very efficient in patients with relapsed or refractory disease[155,156]. Lenalidomide has antiangiogenic and antitumor activity as it downregulates cytokines such as VEGF in CLL patients[157]. However, a clinical trial has reported the appearance of venous thrombosis in approximately 18% of the patients receiving lenalidomide, together with the upregulation of soluble vascular endothelial adhesion molecule 1, tumor necrosis factor-α, and C-reactive protein[158]. Other clinical trials have shown that lenalidomide increases the efficiency of fludarabine and rituximab, although this effect appears to be dependent on specific clinical features of the patients[159-161].
Bevacizumab and VEGF receptor inhibitors
Bevacizumab (Avastin, AVA) is a monoclonal antibody targeting VEGF with proven antiangiogenic properties in multiple hematologic malignancies, such as acute myeloid leukemia, CLL, and non-Hodgkin’s lymphoma[162-164]. Despite its potential relevance in the clinic, the administration of bevacizumab, as a single agent, to CLL patients has shown no significant improvement in clinical trials[165]. However, when bevacizumab was given to CLL patients in combination with other conventional therapies (pentostatin,cyclophosphamide, and rituximab), clinical trials have shown that it prolongs the progression free andtreatment free survival[166]. Vatalanib and pazopanib are potent orally available VEGF receptor tyrosine kinase inhibitors. Both reagents were shown to efficiently induce apoptosis in CLL cells and diminish tumor growth in murine xenograft models[167]. An additional possible avenue to explore VEGF blockage is through epigallocatechin gallate, a green tea extract component that inhibits the VEGFR activation in CLL cells[34,134,168].
Kinase inhibitors and other therapies
Sorafenib is a well-known multikinase inhibitor with proven effective roles in tumor cell signaling,proliferation, and angiogenesis. Sorafenib has been shown to a potent inducer of apoptosis in CLL cells, by a mechanism which involves downregulation of the anti-apoptotic protein Mcl-1[169]. This study also showed that sorafenib was able to overcome the drug resistance effect of stromal cells on CLL cells[169]. Another report has shown that sorafenib blocked the survival signals induced by CXCL12 engagement to its receptor CXCR4, particularly the activation of ERK and MEK kinases, and induced apoptosis in a caspase dependent manner[170]. In another study, sorafenib improved the response of CLL cells to the cytotoxic effect of rituximab or ofatumumab, and this effect involved a decrease in the expression of complement regulatory proteins[171]. It was therefore suggested that sorafenib could constitute a potential second line therapy for refractory CLL patients. Altogether these studies indicate that treatments addressing angiogenic factors and their receptors may constitute a useful approach to treat CLL, particularly when used in combination therapies.
CONCLUSION
Increased bone marrow angiogenesis is a common feature found in CLL, and it is widely accepted that it contributes to the pathogenesis of the disease. CLL cells are active contributors to this aberrant angiogenesis, as they produce and secrete many angiogenic factors and express angiogenic receptors. CLL cells in niches establish functional bidirectional interactions with microenvironmental cells, which induce a proangiogenic profile in CLL cells and enhanced the angiogenic capacity of stromal cells. Clinical trials addressing angiogenic factors have proven to be insufficient as single treatments and indicate that these therapies should rather be used in combined treatments. Moreover, CLL cells in tissues receive survival and proliferation signals that contribute to disease progression. Consequently, the CLL microenvironment is now considered a crucial target for treatment, and current therapies are aimed at the signaling pathways induced by CLL cell-microenvironment interactions.
DECLARATIONS
Authors’ contributions
Contributed to the preparation of the manuscript: García-Pardo A, Redondo-Muñoz J
Availability of data and materials
Not applicable.
Financial support and sponsorship
Work for the author’s laboratory was supported by grants SAF2009-07035, SAF2012-31613, SAF2015-69180-R, PI060400, RD06/0020/0011, RD12/0036/0061 (to García-Pardo A) and SAF2017-86327-R (to Redondo-Muñoz J) from the Ministerio de Ciencia e Innovacion- Fondo Europeo de Desarrollo Regional(FEDER), Madrid; P2010/BMD-2314 from the Comunidad de Madrid/European Union (to García-Pardo A); the Fundación de Investigación Mutua Madrileña (to García-Pardo A); the 2020 Leonardo Grant for Researchers and Cultural Creators (BBVA Foundation) (to Redondo-Muñoz J).
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2021.
杂志排行

Journal of Cancer Metastasis and Treatment的其它文章
- Pancreatic cancer: genetics, disease progression,therapeutic resistance and treatment strategies
- The unclear role of VEGF in POEMS syndrome:therapeutic implications of neoangiogenesis in a rare plasma cell disorder
- The role of immune checkpoint inhibitors in triplenegative breast cancer: recent developments and future perspectives
- Cancer field surgery for locoregional tumor control of cervical carcinoma
- Uterine cervical carcinoma treated with chemoradiotherapy: impact of three-month MRI follow-up on clinical management and outcome
- AUTHOR INSTRUCTIONS