X连锁无丙种球蛋白血症的临床特点及基因诊断
2021-05-09马智丽毛国顺李利
马智丽,毛国顺,李利
(1.安徽医科大学阜阳临床学院,安徽 阜阳236000;2.阜阳市人民医院,安徽 阜阳236000)
X连锁无丙球蛋白血症(X-linked agammaglobulinemia,XLA)为X染色体隐性遗传,主要为男性患病,女性携带,BTK突变为其致病原因,BTK基因全长37.5 kb,编码的蛋白质属于酪氨酸激酶家族,共包含659个氨基酸,发生在BTK基因任意位置的突变均可影响蛋白质功能,造成机体免疫球蛋白缺乏或降低及B细胞分化障碍,造成B细胞显著减少[1],而机体各种免疫球蛋白均是由B细胞分泌的[2],从而使患者自身免疫功下降,发生反复感染,因XLA患者通常保留抗病毒免疫功能[3],所以感染以细菌为主,可累及呼吸道、胃肠道和皮肤等[4]。本研究回顾性分析2017年1月至2018年1月因反复感染最后经基因诊断确诊为X连锁无丙球蛋白血症3例患儿的临床资料,探讨X连锁无丙种球蛋白血症临床表现特点及基因诊断的重要性。
1 临床资料分析
1.1 病例1患儿,男性,1岁9个月,因发热、咳嗽5 d,呕吐1 d,口嘴歪斜2 h入院。患儿10月龄时曾患败血症、臀部脓肿。曾查免疫球蛋白缺如,未予基因检测。
1.1.1 查体T(体温):38.9℃,P(脉率):139次/min,R(呼吸):37次/min,BP(血压):108/71 mmHg,W(体质量):11 kg。神清,精神、营养差,两侧瞳孔等大等圆,对光反射灵敏,右侧鼻唇沟变浅,左侧正常,嘴角稍向左侧倾斜,口唇黏膜正常,咽充血,口腔上颚黏膜可见小溃疡,浅表淋巴结未及肿大,颈项强直,双肺呼吸音正常,未闻及啰音;心率139次/min,心腹(-),右臀部有约2 cm陈旧性瘢痕,臀部肌肉萎缩,左臀部皮肤可见2.5 cm×2.5 cm红肿,触之有硬结,无明显波动感,双下肢肌张力增强,Brudzinski征、Kernig征、Babinski征阴性,四肢末梢暖,CRT:2 s。
1.1.2 辅助检查IgA:0.02 g/L,IgM:0.01 g/L,IgG:0.01 g/L,CD3+:95%,CD4+:57%,CD8+:36%,NK:2%,B细胞:0%。PCT:2.5 ng/mL,CRP:90 mg/L,ESR:20 mm/h,Hb:88 g/L,G实验:38.061 pg/mL。脑脊液常规:白细胞2 000×106,单个核:65%,多个核:35%,葡糖糖:2.93 mmol/L,蛋白:1 311.3 mg/L;EB病毒LgM抗体:阴性,血象、肝肾功能正常,大小便正常。胸片、头颅MR:未见明显异常。
1.1.3 诊断男性,有反复细菌感染病史,实验室检查结果提示免疫球蛋白及B细胞缺乏,无特殊家族史,但根据实验室检查结果,提示免疫缺陷性疾病可能性大,征得家属同意后,委托金域医学检验所对患儿进行基因检测,结果显示,BTK基因发现c.1921C>T突变,此突变使其编码蛋白质的第641位氨基酸残基由Arg(精氨酸)变为Cys(半胱氨酸),为错义突变。
1.1.3 治疗过程根据检验结果及基因结果确诊为XLA,采用抗真菌治疗及丙种球蛋白替代治疗,患儿病情好转出院。
1.2 病例2患儿,男性,1岁,因间断发热4 d入院。
1.2.1 查体T:36.6℃,P:117次/min,R:31次/min,BP:85/55 mmHg,W:8.5 kg。神清,精神可,营养尚可,前囟闭合,口唇黏膜正常,咽充血,扁桃体正常,未触及颈及锁骨上下淋巴结,颈项强直,三凹征阴性,双肺未闻及啰音,心率117次/min,心律整齐,心腹(-),四肢肌张力正常,Brudzinski征、Kernig征、Babinski征阴性,手足无皮疹。
1.2.2 辅助检查IgA:0.05 g/L,IgM:0.20 g/L,IgG:
1.14 g/L,CD3+:97%,CD4+:71%CD8+:25%,NK:2%,B细胞:0%。C反应蛋白37.57 mg/L,血常规基本正常,胸片:两肺纹理增多,紊乱,模糊,提示支气管炎,大小便正常。
1.2.3 诊断患儿既往有多次肺炎病史,根据患儿入院辅助检查提示免疫球蛋白缺乏及外周血B细胞明显降低,不排除免疫缺陷性疾病,取得患儿家属同意后委托武汉圣达医学检验所对患儿及家属进行基因检测,结果显示,该BTK基因发现c.718del(缺失突变),导致氨基酸变化p.Glu240fs(移码突变)。但在该患儿父母双方检测未发现异常,考虑为新发突变,见图1。
1.2.4 治疗过程根据患儿反复感染的临床表现及基因检测,确诊为XLA,予抗生素抗感染及丙种球蛋白替代治疗,患儿好转后出院。
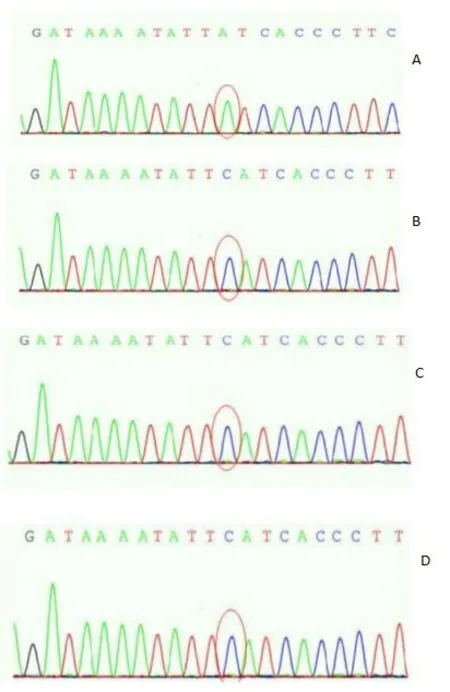
图1 X连锁无丙种球蛋白血症患儿及家属BTK基因检测图谱
1.3 病例3男性,1岁6个月,因反复咳嗽10 d,皮下气胸9 h入院。该患儿既往有反复肺炎病史。且患儿哥哥5个月时因发热死亡,姐姐身体健康。
1.3.1 查体T:36.9℃,P:139次/min,R:41次/min,BP:122/99 mmHg,W:16 kg。神清,精神,营养差,左侧面部肿胀,有握雪感,口唇黏膜正常,咽充血,扁桃体Ⅰ度肿大,颈及锁骨上下淋巴进未及肿大,鼻翼煽动,三凹征阳性,右侧胸壁部局肿胀,有握雪感,两肺可闻及啰音,心率139次/分,心(-),腹软,舟状腹,无包块,肝脾肋下未及,肠鸣音正常,四肢肌张力正常,Brudzinski征,Kernig征,Babinski征阴性,手足无皮疹。
1.3.2 辅助检查IgA:0.03 g/L,IgM:0.15 g/L,IgG:
1.80 g/L,CD3+:94%,CD4+:38%,CD8+:41%,NK:1%,B细胞:2%,PCT:0.5 ng/mL,CRP:82.7mg/L,WBC:24.15×109/L,中性粒细胞比率:78.2%,淋巴细胞比率:14.9%,Hb:113 g/L,G实验,肝肾功能正常,大小便正常。胸部CT:双肺支气管肺炎,有上肺肺大疱伴炎症,两肺明显囊泡状影伴左侧胸腔环状积气征,左侧上胸壁及纵隔气肿。
1.3.3 诊断该患儿既往反复肺部感染病史,且该患儿哥哥于5个月时因发热夭折,再结合患儿临床表现,遗传性免疫缺陷病可能性大,征得患儿家属同意后,委托上海交通大学附属上海儿童医学中心对患儿及父母进行基因检测,结果显示,该患儿BTK基因存在c.906_908del(缺失突变)导致氨基酸改变p.Gly303del,该患儿母亲携带该位点突变,父亲该位点为正常基因,见图2。
1.3.4 治疗过程患儿入院后予抗感染、雾化及吸氧治疗,予丙种球蛋白替代治疗,患儿病情好转后出院。
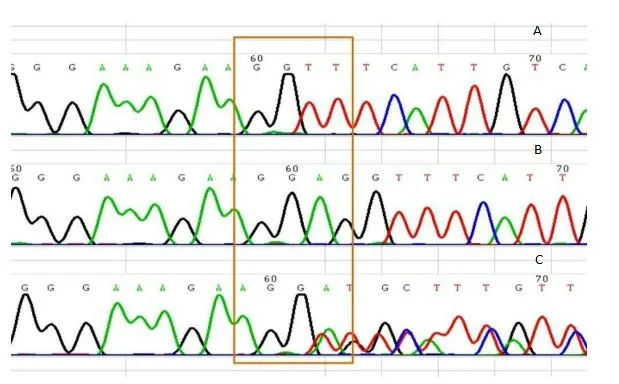
图2 X-连锁无丙种球蛋白血症患儿及家属BTK基因检测图谱
2 结果
3例XLA患儿均为男性,发病年龄为7个月~2岁,3例XLA患儿均有反复感染,表现为败血症、重症肺炎、脑炎、皮肤软组织感染等。实验室结果提示免疫球蛋白缺乏及外周血B细胞缺如或明显降低。3例XLA患儿最后经基因均确诊为XLA,均存在BTK基因突变,分别为BTK基因的错义突变、移码突变及碱基缺失。
3 讨论
XLA是X染色体隐性遗传病,患者通常为男性,易反复感染,且以细菌感染为主,以血清免疫球蛋白降低、B细胞缺如或降低为主要临床表现,通常在母源性抗体从婴儿体内消失后开始出现反复感染,即通常在6个月后开始出现症状,但也有报道称25%的XLA患儿在4个月前会出现症状[5],本研究中3例患儿均为男性,首发时间均在1岁内,均有反复感染病史,表现为败血症、重症肺炎、脑炎、皮肤软组织感染等。实验室结果均提示,免疫球蛋白缺乏及外周血B细胞缺如或明显降低,与XLA临床特点相符。3例患儿虽均有反复感染,但感染程度和部位存在差异,其中病例1患儿10个月时发生败血症和臀部脓肿,1岁左右时又确诊为隐球菌脑膜炎;病例3患儿确诊时间较晚,但之前一直有反复肺部感染,且感染症状较重;而病例2患儿较前两位患儿相比,仅表现为发热,且感染症状不重。最终3例患儿均经基因确诊为XLA,均存在BTK基因突变,但突变的类型不同,分别为错义突变、移码突变及碱基缺失。有文献[6]指出XLA患儿突变基因型与临床表现有关,本研究中3例患儿基因突变类型不同,感染程度也存在差异,可能与基因型及临床表型有一定相关性,但本研究本例数较少,有文献[7]指出,相同基因突变在不同个体表型也不同,具体表型和基因型间的关联仍需进一步深入研究。其中病例1BTK基因存在c.1921C>T突变,为错义突变,但因该患儿父母未接受基因检测,因此,该突变来源未明确;病例2BTK基因存在c.718del(缺失突变),但在该患儿父母双方检测未发现异常,考虑为新发突变;病例3BTK基因存在c.906_908del(缺失突变),该患儿母亲携带该位点突变,父亲该位点为正常基因,从而确定患儿人致病基因来源于母亲,符合X染色体隐性遗传。从研究中可发现虽然XLA为遗传性疾病,但本研究中例2患儿为新发突变,无家族史,父母中也未检测到突变基因,这与相关报道认为有30%~50%的XLA患儿无家族史[8]相符。
目前XLA的治疗仍以规律应用IVIG为主,有皮下应用和静脉应用两种方式,一般起始剂量为每月400~600 mg/kg。静脉应用为每3~4周1次,皮下应用为每周或隔周1次,因个体差异较大,具体间隔时间和剂量应根据患儿的实际情况,使IgG的浓度>5 g/L[9]。虽然应用IVIG可明显降低患儿的感染率,但长期应用IVIG,有感染血液传播性疾病的可能,如乙肝、丙肝、HIV,也有研究者认为,长期应用IVIG的患者有发生直肠癌及B淋巴细胞白血病的可能[10-11]。
综上所述,XLA作为一种免疫性缺陷病,早期发现和诊断具有重要意义,应提高临床医生对该病的认知,在临床工作中发现反复感染及免疫球蛋白明显降低的患儿应及时完善基因检查,早期干预治疗可显著提高患儿的生存质量,延长生存期。
