UPLC-MS/MS法测定鸡蛋中17种硝基咪唑类药物及代谢物
2021-05-08尹青春谭高好邓浩潘永波张容鹄徐彬
尹青春 ,谭高好,邓浩,潘永波,张容鹄,徐彬
1.海南省农业科学院农产品加工设计研究所,海南省热带果蔬冷链研究重点实验室(海口 570100);2.海南省食品检验检测中心(海口 570100)
硝基咪唑类药物是一类具有5-硝基咪唑环共同结构的合成抗菌类药物,常见的有甲硝唑、洛硝哒唑、地美硝唑、替硝唑、羟甲基甲硝咪唑等。硝基咪唑类衍生物易与生物体内的许多大分子如酶、DNA和受体等发生超分子结合[1],在抗癌[2-3]、祛痘[4]、抗厌氧菌[5]、抗寄生虫[6]等方面有广泛医学用途。此外,使用此类药物对动物体重还具有增加作用[7],因此在畜禽养殖中被广泛违法使用。有研究表明,硝基咪唑类药物和其代谢物具有致突变和致癌性[8],禽类肉和蛋制品中的硝基咪唑类药物残留对消费者健康构成直接威胁。
我国和欧盟已将硝基咪唑类药物列为动物性食品不得检出的对象。GB 31650—2019《食品中兽药最大残留限量》[9]及《农业部公告第235号》[10]明确规定地美硝唑、甲硝唑允许作治疗用,但不得在动物性食品的所有可食组织中检出。欧盟理事会也将硝基咪唑类药物列入A类禁用药物,不得在任何食品中检出[11]。但违法使用硝基咪唑类药物现象屡禁不止,鸡蛋等农产品抽检不合格仍时有发生。
超高效液相色谱-串联质谱(UPLC-MS/MS)法具有快速、高效、高特异性、高灵敏度等特点,是目前检测硝基咪唑类药物残留的常用方法。李小桥等[12]使用UPLC-MS/MS法测定牛肉中4种硝基咪唑类药物残留量。鸡蛋样品中多种硝基咪唑类药物及其代谢物的UPLC-MS/MS批量检测方法尚未见报道。此次试验从提取溶剂、净化方式、复溶液、液相流动相、色谱柱、质谱条件等参数进行优化,建立UPLC-MS/MS方法,同时测定鸡蛋中17种硝基咪唑类药物及其代谢物,为政府监管部门提供技术参考。
1 材料与方法
1.1 仪器与试剂
Thermo TSQ Quantiva三重四极杆质谱仪[配有电喷雾离子(ESI)源]、Ultimate 3000超高效液相色谱仪,美国Thermo公司;Centrifuge 5804 R高速冷冻离心机(Eppendorf公司);多管涡旋混合器(德国Heidolph公司);Mettler XS204型十万分之一分析天平(美国Mettler Toledo公司);SCIENTZ-950E超声波提取器(宁波新芝生物科技公司)。
16种硝基咪唑混标(100 μg/mL,A ChmTek.Inc.),其它均为色谱纯。鸡蛋购于海口市秀英区秀英街道向荣村农贸市场。
1.2 试验方法
1.2.1 标准工作溶液的配制
准确称取适量羟甲基甲硝咪唑、罗硝唑D3标准品,配制成质量浓度为100和1.0 μg/mL的标准使用溶液;17种混合标准使用溶液(1.0 μg/mL):准确吸取100 μg/mL 16种硝基咪唑混标和100 μg/mL的羟甲基甲硝咪唑标准使用溶液各100 μL,用甲醇定容至10 mL。所有标准使用溶液置于-18 ℃冰箱冷藏保存。
1.2.2 样品前处理
将鸡蛋样品混合均匀并准确称取2.0 g,加入100 μL质量浓度为1.0 μg/mL的罗硝唑D3的内标标准使用溶液,再加入10 mL 1%甲酸乙腈,漩涡10 min,超声10 min,以10 000 r/min离心5 min;净化:取2.0 mL上清液,加入200 mg C18,漩涡10 min,以10 000 r/min离心5 min,取1.0 mL上清液,于40 ℃氮吹干,加入1.0 mL 50%甲醇水溶液复溶,过0.22 μm有机相膜,上机测定。
1.3 仪器条件
色谱柱,ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm);流动相A,0.1%甲酸水溶液;流动相B,甲醇溶液;柱温35 ℃;进样量5.0 μL,流速0.3 mL/min。梯度洗脱程序:0~0.25 min,5% B;0.25~3 min,5% B~65% B;3~4 min,65% B~99% B;4~5 min,99% B;5~5.01 min,99% B~5% B;5.01~7 min,5% B。
质谱:离子源ESI源,正离子模式;扫描模式:选择反应监测(SRM)模式;喷雾电压3.5 kV;汽化温度350 ℃;离子传输温度 350 ℃;鞘气(氮气)压力32 Arb;辅助气(氮气)压力5 Arb;碰撞气:高纯氩气,压力为1.5 mTorr。优化后17种硝基咪唑类药物及其代谢物的质谱参数见表1。
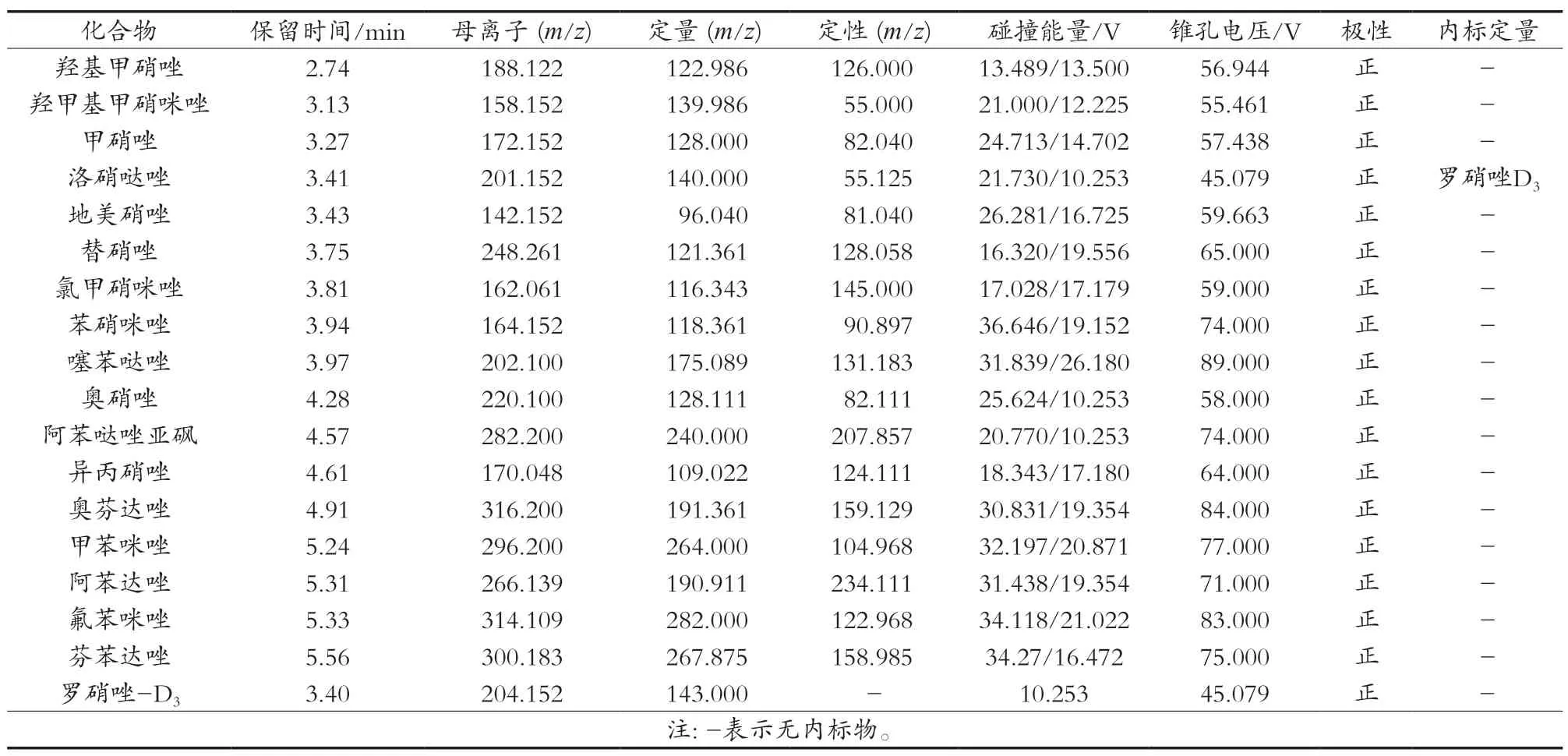
表1 17种硝基咪唑类药物及其代谢物的质谱参数
2 结果与讨论
2.1 色谱柱的筛选
试验对比了Waters ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm)、Thermo Accucore C18(2.6 μm,2.1×100 mm)和ACE Ultra Core 2.5 Super C18(2.5 μm,2.1 mm×100 mm)3种色谱柱对17种硝基咪唑类药物及其代谢物的分离效果。图1为Waters BEH C18色谱柱的目标化合物总离子色谱图,其分离效果最好。刘燕等[13]在研究超高效液相色谱-四级杆-静电场轨道阱高分辨质谱联用(UPLC-Q-Exactive Orbitrap)快速检测饲料中16种硝基咪唑类药物时也发现Waters BEH C18色谱柱对16种硝基咪唑类药物的检测灵敏度及色谱峰型较好。因此选择Waters BEH C18进行方法研究。

图1 目标化合物总离子色谱图(BEH柱)
2.2 流动相的筛选
试验对比了0.1%甲酸水/甲醇、5 mmol/L乙酸铵水溶液(含0.1%甲酸)/甲醇、0.1%甲酸水/乙腈、5 mmol/L乙酸铵水溶液(含0.1%甲酸)/乙腈4种不同流动相体系对17种硝基咪唑类药物及其代谢物的分离及响应效果。结果表明:体系含有甲醇的流动相对大部分硝基咪唑类药物及其代谢物的分离效果均较好,其中0.1%甲酸水/甲醇的色谱峰型、信号响应、稳定性最优。周兴鑫[14]在建立生鲜乳中250种兽残高通量快速筛查平台时发现流动相中加入0.1%的甲酸对于各化合物响应强度具有明显的提高。因此选择0.1%甲酸水/甲醇作为体系的流动相。
2.3 样品提取液的筛选
分别以甲醇、乙腈、1%乙酸乙腈、1%甲酸乙腈、5%甲酸乙腈等11种有机溶剂作为提取液,对比17种硝基咪唑类药物及其代谢物的提取效果。结果表明:使用1%甲酸乙腈和5%甲酸乙腈提取时,回收率为70%~130%,且1%甲酸乙腈和5%甲酸乙腈提取效果无明显差异,考虑到绿色环保,选择1%甲酸乙腈作为提取液。
2.4 净化方式的筛选
已有文献报道采用Waters PRIME HLB、MCX[15]、PCX[16]、RETAIN-CX等作为净化样品的固相萃取柱,还有采用PSA[17-18]、MgSO4、C18或者几种混合作为净化样品的吸附剂。基于以上文献报道,试验比较了MCX、HLB、PSA、C184种净化方式的净化效果,以加标量为50 μg/kg的回收率为指标,净化效果最好的是Agela Cleanret S C18,目标物回收率达90%~110%,其他3种净化方式回收率浮动较大。因此,采用200 mg Agela Cleanret S C18吸附剂进行样品净化。
2.5 复溶液的选择
取1 mL净化后的提取液,经氮气吹干浓缩后,对比不同比例的乙腈和甲醇作为复溶液对目标物回收率的影响。结果显示:使用1%乙腈水、5%乙腈水作为复溶液时,大部分目标物的峰型较差;使用10%甲醇水作为复溶液时,小部分目标物的峰型不好;使用50%和80%甲醇水溶液时,各目标物峰型较好,且均可获得较为满意的回收率。考虑到成本,选择50%甲醇水溶液为复溶液。
2.6 线性范围、检出限、定量限、精密度
采用优化好的UPLC-MS/MS参数进行方法学研究,结果如表2所示。17种硝基咪唑类药物及其代谢物均呈现良好的线性关系,线性范围为1.0~100 ng/mL,相关系数r≥0.999 0,羟基甲硝唑、羟甲基甲硝咪唑、甲硝唑、洛硝哒唑、地美硝唑、替硝唑、氯甲硝咪唑、苯硝咪唑、噻苯哒唑、奥硝唑、阿苯哒唑亚砜、异丙硝唑、奥芬达唑、甲苯咪唑、阿苯达唑、氟苯咪唑和芬苯达唑的检出限分别为0.056,0.115,0.065,0.046,0.026,0.088,0.115,0.032,0.016,0.083,0.010,0.016,0.004,0.006,0.005,0.010和0.003 μg/kg,定量限均为5.0 μg/kg;SN/T 2624—2010[19]规定硝基咪唑类测定低限为1.0 μg/kg;SN/T 1626—2019[20]规定羟甲基甲硝咪唑测定低限为1.0 μg/kg,甲硝唑、洛硝哒唑、地美硝唑、替硝唑、奥硝唑测定低限均为0.5 μg/kg。方法的检出限均低于标准方法检测低限,说明该检测方法灵敏度较高。对质量浓度为10.0 ng/mL的17种硝基咪唑类药物及其代谢物的标准溶液进行6次重复测定,该方法的δRSD(n=6)为0.4%~2.3%,说明仪器具有良好的精密度。
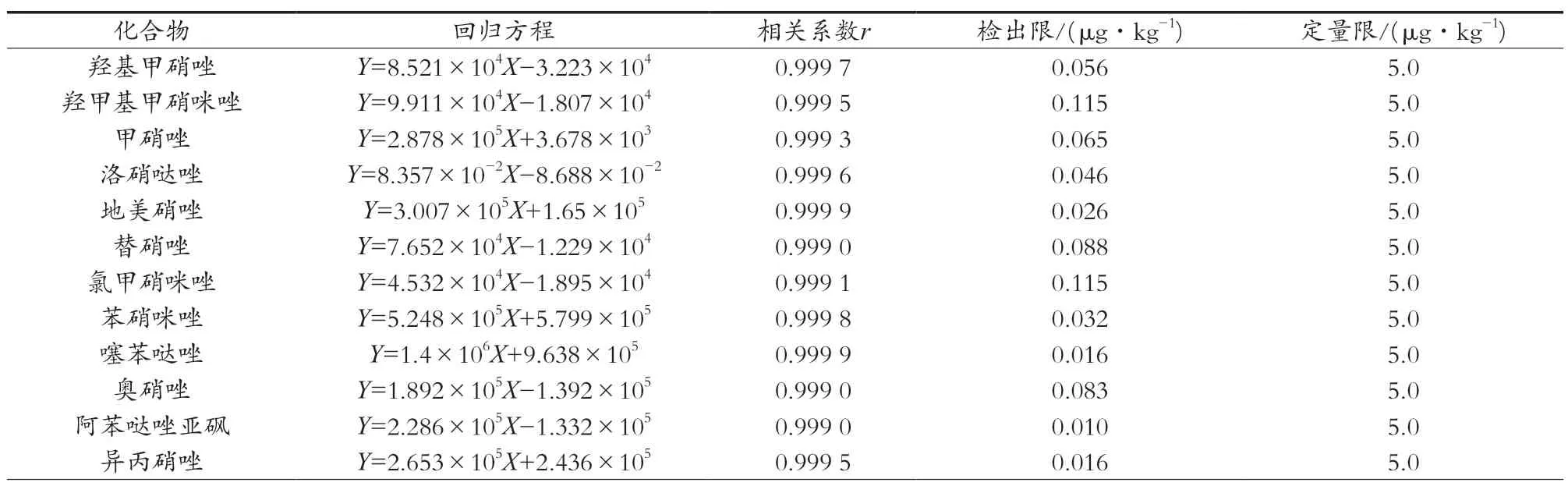
表2 17种硝基咪唑类药物及其代谢物的线性关系、检出限、定量限

接表2
2.7 回收率试验
将17种目标化合物添加到空白鸡蛋样品进行50,100和250 μg/kg 3个水平的添加回收试验,每个水平取6个平行样,平均回收率为73.1%~117.7%,方法的相对标准偏差δRSD为0.6%~6.5%,说明该方法回收率及重复性较好。
2.8 样品测定
采用试验建立的UPLC-MS/MS方法对海南省18个市县菜市场抽检的鸡蛋样品共78批次中17种硝基咪唑类药物及其代谢物的残留量进行测定,每个样品平行测定2次,结果均未检出此类兽药。虽然,目前在抽检的鸡蛋中均未检出17种硝基咪唑类药物及其代谢物,但仍需加强此类兽药残留检测的监控。
3 结论
通过优化分离条件和样品提取净化条件,建立了UPLC-MS/MS内标法测定鸡蛋中17种硝基咪唑类药物及其代谢物的方法。样品经1%甲酸乙腈提取,经C18吸附剂净化,以ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm)色谱柱分离,0.1%甲酸水溶液-甲醇作为流动相梯度洗脱,电喷雾正离子模式(ESI+),多重反应监测(MRM)扫描,洛硝哒唑内标法定量,其他16种硝基咪唑类药物及其代谢物外标法定量。结果显示,17种目标化合物在1.0~100 ng/mL范围内呈良好的线性关系,相关系数r≥0.999 0,在3个不同浓度添加水平下,平均加标回收率为73.1%~117.7%,方法δRSD为0.6%~6.5%,检出限为0.003~0.115 μg/kg,定量限为5.0 μg/kg。该方法前处理简单,具有净化效果好、准确快速的特点。测定海南省18个市县抽检的鸡蛋样品(共78批次),结果均未检出此类兽药。该方法适用于大批量鸡蛋中17种硝基咪唑类药物及其代谢物的快速筛查、确证及定量。
