1,2-二(2-甲氧基苯基)-1,2-乙二胺的制备方法研究
2021-05-06邸相杰黄晴菲王启卫
邸相杰, 邹 胜, 黄晴菲*, 范 挺*, 王启卫*
(1. 中国科学院 成都有机化学研究所,四川 成都 610041; 2. 中国科学院大学,北京 100491)
手性1,2-二芳基-1,2-乙二胺作为一种优势的手性配体或催化剂,广泛应用于不对称合成反应之中。该配体不仅可以和多种金属,如Ru、Rh、Ir等配位,用于不对称催化氢化[1-3]、转移氢化[4-9]、Diels-Alder反应[10]及Heck反应[11],还可以用于合成结构多样的有机胺类催化剂用于催化醛、酮等羰基化合物的串联环化反应[12]。其中,手性1,2-二(2-甲氧基苯基)-1,2-乙二胺作为一种手性金属配体[3],或有机小分子催化剂的前体化合物,以其独特的空间效应和电子效应,在不对称氢化[8]和Betti/aza-Michael反应[12]中表现出良好的催化效果。
Scheme 1
Scheme 2
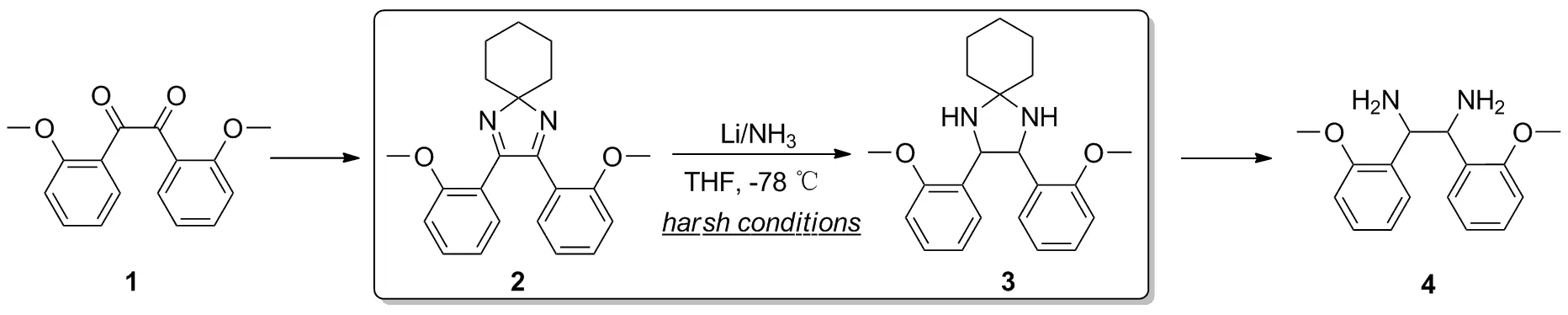
目前,1,2-二芳基-1,2-乙二胺主要是通过以N-亚苄基-1,1,1-三甲基硅胺[13]、二苯基乙二醇[14]或二苯基乙二酮[1]为起始原料制备得到的。通过消旋体拆分的方法能得到光学纯度的手性乙二胺衍生物。然而,也正是由于甲氧基所处的位置及较强的给电子共轭效应的影响,直到2004年,Carpentier等人才以二(2-甲氧基苯基)乙二酮1为原料,经过胺化、还原、水解等步骤完成了1,2-二(2-甲氧基苯基)-1,2-乙二胺的合成(Scheme 1)。但在其还原亚胺的关键步骤中,要使用金属锂/液氨在-78 ℃的反应条件下进行,其在工业中的放大反应受限。因此,寻找一种条件更为温和的还原亚胺的方法,不仅能够满足1,2-双(2-甲氧基苯基)-1,2-乙二胺的合成及下游的手性物质的合成需要,也可以为其工业化生产提供必要的方法学参考。
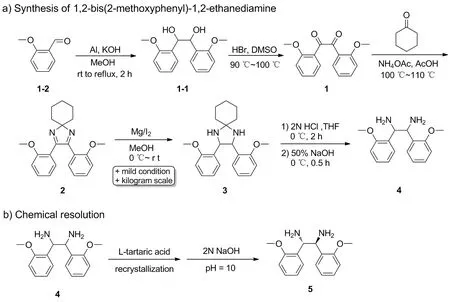
本文以廉价易得的邻甲氧基苯甲醛为原料,着力解决亚胺中间体还原过程中反应条件苛刻等问题,首次在室温下,利用Mg作为还原剂,在催化量碘单质的引发下,以93%的分离收率得到关键中间体3。该制备方法不仅后处理简单高效,同时,在放大制备的条件下,该关键步骤的收率也可保持不变。随后,以L-酒石酸对其进行拆分得到光学纯的(S,S)-1,2-二(2-甲氧基苯基)-1,2-乙二胺(Scheme 2),并通过1H NMR,比旋光度和手性HPLC对手性乙二胺的结构及纯度经进行了确认。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE 300 MHz型核磁共振仪(TMS为内标);LC-20AT VP型高效液相色谱仪;Polarimeter 341型旋光仪。
邻甲氧基苯甲醛,98%,阿法埃莎(中国)化学有限公司;48%氢溴酸,98%环己酮,成都市科隆化学品有限公司;其余所用试剂均为分析纯。
1.2 合成
(1)1-1的合成
取邻甲氧基苯甲醛(50.02 g, 0.37 mol, 1 eq.)和铝箔(19.85 g, 0.74 mol, 2 eq.)加入2 L反应釜中,加入100 mL甲醇,搅拌下加入KOH(123.32 g, 2.20 mol, 6 eq.)的甲醇(450 mL)溶液,缓慢升温至回流,反应6 h。冷却至室温,过滤,滤饼用甲醇(3×20 mL)洗涤,减压蒸去甲醇,残留物缓慢倒入1 L水中,剧烈搅拌1 h;过滤,滤饼用水(3×20 mL)洗涤,于50 ℃真空干燥得白色块状中间体1-148.03 g,无需进一步纯化,直接进行下一步反应。
化合物1-1[15]: m.p.151~153 ℃;1H NMR(300 MHz, CDCl3)δ: 7.26~7.15(m, 4H), 6.91~6.74(m, 4H), 5.26(d,J=5.0 Hz, 2H), 3.69(d,J=10.6 Hz, 6H), 3.13(d,J=5.4 Hz, 2H)。
(2)1的合成[16]
将1-1(48.03 g)溶于175 mL DMSO中,搅拌下滴加48%氢溴酸(75 mL),升温至内温90~100 ℃,反应3 h(TLC检测)。反应结束后,剧烈搅拌下,趁热倒入500 mL水中,搅拌10 min,静置过夜;倾出上清液,收集沉淀,于50 ℃真空干燥得深褐色针状固体152 g, m.p.130~131 ℃;1H NMR(300 MHz, CDCl3)δ: 8.08(t,J=1.3 Hz, 2H), 7.55(ddd,J=1.8 Hz, 7.4 Hz, 9.1 Hz, 2H), 7.13(dd,J=0.8 Hz, 7.7 Hz, 2H), 6.95(d,J=8.3 Hz, 2H), 3.57(s, 6H)。
(3)2的合成[17]
将1(52 g)溶于175 mL冰醋酸中,搅拌下加入NH4OAc(98.91 g, 1.28 mol),加入环己酮50 mL,加热至内温为100~110 ℃,反应2 h(TLC检测)。搅拌下将反应液趁热倒入500 mL水中,搅拌10 min;静置后倾出上清液,再用300 mL水洗涤沉淀,用乙酸乙酯(150 mL)重结晶得黄色固体227.76 g,收率56%, m.p.110~112 ℃;1H NMR(300 MHz, CDCl3)δ: 7.49(d,J=7.4 Hz, 2H), 7.30(m, 2H), 7.00(m, 2H), 6.71(d,J=8.3 Hz, 2H), 3.15(s, 6H), 1.97(m, 4H), 1.84(m, 4H), 1.73(m, 2H)。
(4)3的合成
取化合物2(27.76 g, 0.10 mol, 1 eq.),加入甲醇540 mL,冰水浴下,先向反应器中加入3.7 g镁屑和碘粒(0.01 g, 0.052 mol, 0.0005 eq.),待气泡缓和后,每1 h加入镁屑(8.4 g,共计37.11 g),加毕,自然升至室温,搅拌反应过夜。反应结束后,将反应液用硅藻土过滤,旋干滤液,残留物加入二氯甲烷250 mL及水250 mL,调至pH=7,分液,水相用二氯甲烷(2×50 mL)萃取,合并有机相,无水硫酸钠干燥,过滤,滤液旋干得化合物326.08 g,收率93%, m.p.65~66 ℃;1H NMR(300 MHz, CDCl3)δ: 7.23~7.11(m, 4H), 6.86~6.76(m, 4H), 4.53(s, 2H), 3.62(s, 6H), 2.56(s, 2H), 1.80(m, 4H), 1.72(m, 4H), 1.47(t,J=5.6 Hz, 2H)。
(5)4的合成
取化合物3(26.08 g),加入四氢呋喃120 mL,冰水浴冷却,缓慢滴加 2N HCl(83 mL),滴毕,于室温搅拌反应1 h。加入纯水(138 mL),用二氯甲烷(3×50 mL)萃取,合并水相,用2N NaOH调至pH=10,用二氯甲烷(3×60 mL)萃取,合并有机相,无水硫酸钠干燥,过滤,滤液旋干得化合物417.75 g,收率82%, m.p.84~86 ℃,dr3/1;1H NMR(300 MHz, CDCl3)δ: 7.23~7.20(m, 2H), 7.14~7.09(m, 2H), 6.84~6.79(m, 2H), 6.77~6.75(m, 2H), 4.45(s, 2H), 3.78(s, 6H), 1.80(s, 4H);1H NMR(300 MHz, CDCl3)δ: 7.30~7.28(m, 2H), 7.23~7.20(m, 2H), 6.95~6.86(m, 2H), 6.84~6.79(m, 2H), 4.50(s, 2H), 3.76(s, 6H), 1.80(s, 4H)。
(6)5的化学拆分[18]

2 结果与讨论
2.1 反应条件的筛选
对反应条件进行了优化探索。出于金属单质的性质及成本考虑,首先尝试使用金属镁代替锂直接还原亚胺,结果显示该反应无法进行(Entry 1)。基于文献[13,19]报道,考虑到该还原过程可能为金属介导的自由基还原。在室温下采用甲醇作为质子源,对引发剂进行了考察(Entry 2~3)。结果发现碘单质及AIBN均能够推动还原反应的进行。基于成本的考虑,以碘单质作为添加剂,对金属镁的用量进行了筛选,当用量增大到15 eq.时(Entry 5),能够以93%的收率得到目标化合物,继续增加金属镁的用量,对收率提高不明显(Entry 6)。随后,进一步对碘的用量进行了筛选,结果发现碘的用量几乎对反应收率无影响(Entry 7~8)。

表1 亚胺还原的条件筛选Table 1 Condition screening for imine reduction
2.2 反应放大
为进一步考察反应的实用性,将还原反应进行了放大。结果显示,在室温下,将反应在放大至0.5 kg时,收率依然较高。此外,该过程的反应单一,后处理简单,未反应完的镁屑通过简单的活化即可回收使用。同时,该反应能够在空气氛围下进行。

表2 亚胺还原的放大Table 2 Large-scale of imine reduction
以廉价易得的邻甲氧基苯甲醛为原料,首次在室温下,以Mg为还原剂,以93%的分离收率得到关键中间体(3)。通过对该关键步骤的放大实验,进一步验证了该方法的应用潜力。
