苄基位C—H键胺基化研究进展
2021-05-06陈俊多郭右安郭长彬
陈俊多, 郭右安, 郭长彬
(首都师范大学 化学系,北京 100048)
1 苄基位C-H键胺基化简介
胺类化合物在天然产物、医药、农药、功能有机分子等方面有广泛应用。在2016年最畅销药品前200名中,85%含有C—N键。其中,苄基胺作为一种重要结构单元,广泛存在于多种抗菌、抗癌、抗抑郁、抗阿尔茨海默病药物中。C—H键的直接官能化区别于传统偶联反应需要对底物进行卤化等预处理,底物无需预官能化,更少的步骤可以减少底物浪费和副反应发生,近年来已成为热门研究领域,并且还在向着反应多样化、底物多样化、高产率、高选择性、低成本等方向发展。其中C(sp2)—H键直接官能化研究比较成熟,与之相比,C(sp3)—H键直接官能化研究相对滞后。然而,苄基位碳原子位置特殊,反应中形成的游离基、碳正离子等中间体可与芳环形成p-π共轭,稳定了中间体和过渡态,有利于反应进行。因此苄基位C—H键胺化凭借结构优势和良好的应用前景,在C—H键官能化领域中受到广泛关注。
Scheme 1

Scheme 2

Scheme 3

Scheme 4
苄基位C—H键胺化的研究重点关注以下3个方面:(1)氮源问题,不同氮源的物化性质会影响其应用,叠氮化物是最常见的氮源[1-6],此外还有胺类[16-18]、重氮盐[24]、二噁唑酮[28-29]等作为氮源,其制备、保存、分离、操作的难易都需要考虑;(2)立体选择性问题,苄基位在胺化后通常为变为手性碳,手性化合物在医药方面有着独特的性质,因此如何提高立体选择性是一个很有意义的问题;(3)区域选择性问题,很多药物分子通常存在多个不同活性的反应位点,能否顺应或克服反应位点的先天反应倾向,实现对指定位置的选择性胺化也是有价值的问题。选择性问题通常解决思路是设计含大位阻基团的手性催化剂,利用空间排斥作用和其他相互作用来控制反应的选择性。除此之外还有诸如氮原子的保护,清洁廉价催化剂的开发等都是值得研究优化的方向。
含氮杂环应用广泛,是一类重要的有机物。分子内C—H键胺化是构建含氮杂环的有力路径,C—H键胺化报道中目标产物也多为含氮杂环,分子间胺化的报道偏少。本文筛选近5年报道中涉及苄基位C—H键胺化的例子,突出介绍其在催化方式、反应机理、反应条件、底物、选择性等方面的创新和优势。
2 氮源
2.1 以叠氮化物为氮源的苄基位C—H键胺化
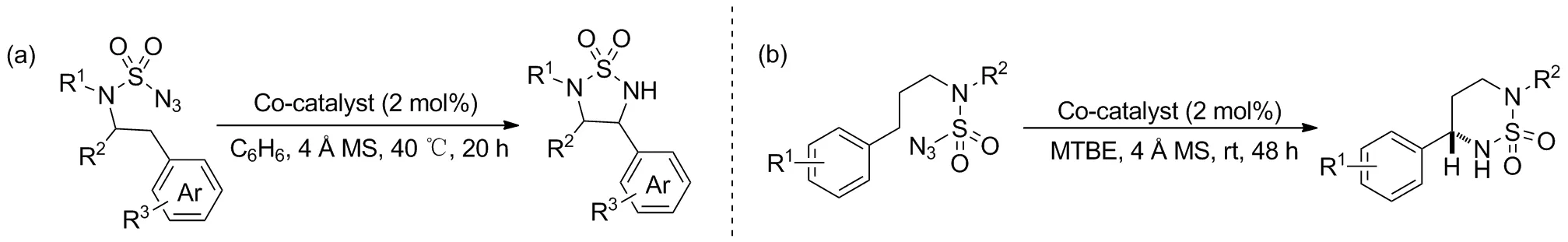
叠氮化合物价格低廉,是最常被用作C—H键胺化氮源的物质之一。2016年Lu等[1]报道了钴催化分子内C(sp3)-H胺化的方案(Scheme 1a)。以磺酰叠氮化物为底物,利用金属自由基催化策略,设计合成并筛选出最佳的钴-卟啉自由基催化剂。在反应开始时激活底物脱氮气分子得到氮自由基,随后发生1,5-氢原子转移(HAT)活化远端C(sp3)—H键进行环合得到五元环磺酰胺衍生物。该方案无需加入其他添加剂,催化剂可直接购买后修饰得到。底物范围广,其中苄基位C—H键的催化胺化对不同N-取代、芳环取代及其他杂芳环均耐受,产率80%~95%。之后,该团队[2]又尝试了应用钴-卟啉自由基催化剂催化分子内苄基位胺化合成六元环(Scheme 1b),用于制备六元环磺酰胺衍生物。、一系列苄基型底物产率89%~94%且ee值高于90%。与上文报道区别在于底物被活化形成自由基之后通过1,6-HAT活化指定位置C—H键,再与原位氮原子作用环合胺化;产物具有手性,简单后处理可以得到手性衍生物。
2017年,Ma等[3]报道了铜催化C—H键胺化的方法(Scheme 2),用于构建三取代咪唑环。
Scheme 5
Scheme 6

Scheme 7
Scheme 8
Scheme 9
氮源叠氮基三甲基硅烷进攻底物N-苄基烯胺的双键得到亚胺自由基,经历1,5-HAT活化苄基位C—H键,最后与亚胺环合得到2,4,5-三取代咪唑,其中4-位被三氟甲基取代,此类芳香化合物在医药上有重要应用。苄基型底物范围广泛,收率30%~85%。反应体系对N-烷基、烯基、其他杂芳基取代也可顺利进行,产率良好。
2017年,Alt等[4]报道了铁催化C(sp3)—H胺化的例子(Scheme 3)。该方案以邻烷基苯基叠氮化合物作为唯一底物,铁配合物催化剂先与叠氮基作用,随后通过HAT诱导指定位置C—H键活化,最后环合。该方案通用性良好,对各种取代基均具有耐受性,苄基型底物可顺利完成胺化得到取代吲哚啉。当底物含多个反应位点时,会发生1,5-或1,6-HAT从而得到取代吲哚啉和四氢喹啉的混合物,遗憾的是报道中没有对两种产物的选择性做出优化。
2018年,Shing等[5]开发了一条具有轴向N-杂环卡宾配体的铁(III)卟啉催化剂用于催化各种烷基叠氮化物的δ-位C(sp3)—H胺化得到一系列吡咯烷衍生物(Scheme 4)。反应通过微波加热,在半小时内完成,当δ-位为苄基位时产率90%。报道侧重于探究其通用性而未拓展更多苄基型底物反应性。对于碳链更长的底物,调控反应条件还可实现选择性胺化ε-位C—H键合成哌啶,是构建结构复杂环系的一种有吸引力的方法。
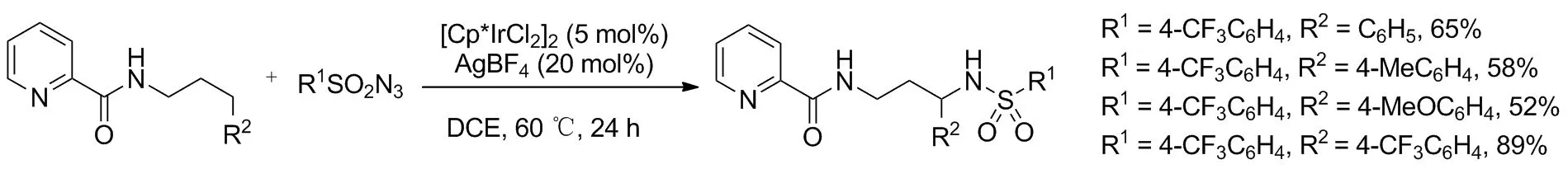
2016年,Xiao等[6]首次报道了铱(Ⅲ)高区位选择性催化C(sp3)—H胺化的方案(Scheme 5)。以一系列N-直链或支链烷基取代的吡啶甲酰胺为底物,对甲基苯磺酰叠氮化合物为氮源,于60 ℃反应24 h胺化酰胺γ-位亚甲基C—H键,得到γ-磺酰胺取代吡啶甲酰胺。底物范围广泛,γ-位被芳基取代时,苄基位C—H顺利胺化,产率52%~89%。机理研究表明,铱催化剂活化后与底物两个氮原子配位,氮源氧化加成至铱中心,随后铱原子上的配体帮助铱-氮烯选择性插入γ-位C—H键完成胺化。
2.2 以磺酰胺类化合物为氮源的苄基位C—H键胺化
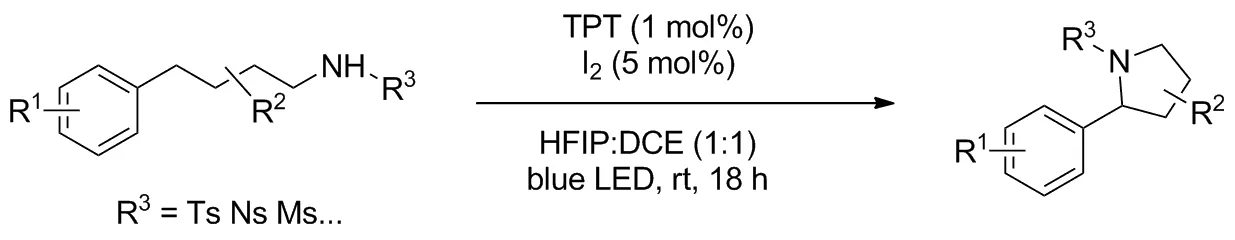
磺酰胺类化合物相比叠氮化合物更加安全,也方便后续处理,所以以此作为氮源对苄基位C—H键进行胺化的报道也很多。2017年,Becker等[7]报道了一种前所未有的利用碘与光催化剂协同催化的苄基位C—H键胺化反应(Scheme6),用于合成吡咯烷。碘单质活化后与底物氮原子作用释放自由基,经历1,5-HAT活化并碘化远端苄基位,最后脱去一分子碘化氢环合得到目标产物。而光氧化还原催化剂2,4,6-三苯基吡喃四氟化硼盐(TPT)则在光照下氧化碘化氢重新使碘单质再生。反应无需大量碘参与,且底物无需碘化预处理。反应体系官能团耐受性良好,产率31%~96%,大多数高于80%。2018年该课题组[8]使用了极少被报道的溴化物催化C(sp3)—H胺化的例子(Scheme 7)。以N-取代磺酰胺为底物,以四丁基溴化铵为催化剂,间氯过氧苯甲酸为氧化剂,分子内胺化远端C(sp3)—H键得到吡咯烷。底物范围广,苄基型底物产率68%~98%。反应可耐受苯环及氮原子上各种取代基,对其他杂芳环也可顺利进行,最高产率接近定量。机理研究中成功分离出N-溴化中间体,为以后溴催化剂的探索提供指南。
Scheme 10

Scheme 11

Scheme 12
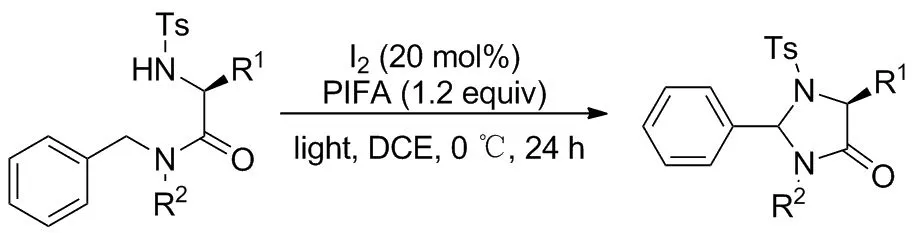
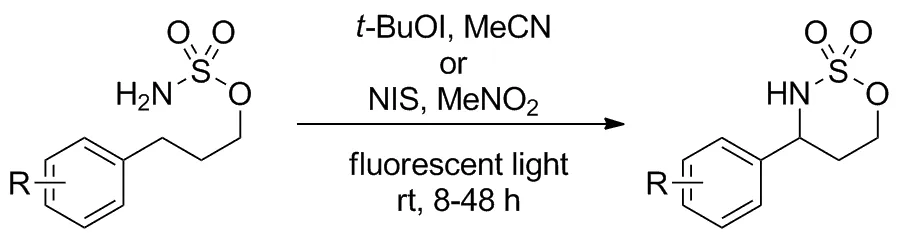
2019年,Kanyiva等[9]报道了碘催化氨基酸衍生物的C(sp3)—H/N—H偶合不对称合成4-咪唑啉酮的方案(Scheme 8)。受Suárez[10]和Fan[11]启发,采用高价碘在光照条件下反应,无需过渡金属,温和的反应条件为多种官能团提供保护。反应开始碘单质与高价碘试剂PIFA生成活性三氟乙酰基碘,与底物磺酰胺氮原子作用释放氮自由基中间体和碘自由基,邻位磺酰基起到了稳定作用;随后发生分子内HAT,活化指定碳原子并与碘自由基成C—I键;碘原子经历氧化加成-还原消除得到环化目标产物,苄基位胺化产率普遍在80%以上。相似地,同年Kiyokawa课题组[12]报道了一种碘催化的以氨基为端基的磺酰胺类底物完成分子内胺化的方案(Scheme 9),用于合成含硫杂环。并且筛选出两组催化体系:一组以叔丁氧基碘为氧化剂,乙腈为溶剂;另一组以N-碘代丁二酰亚胺(NIS)为氧化剂,硝基甲烷为溶剂,两组条件对不同类型底物反应性不同,共同表现出该方案广泛的底物范围,对苯环上连有各种取代基的苄基型底物产率均在80%左右。
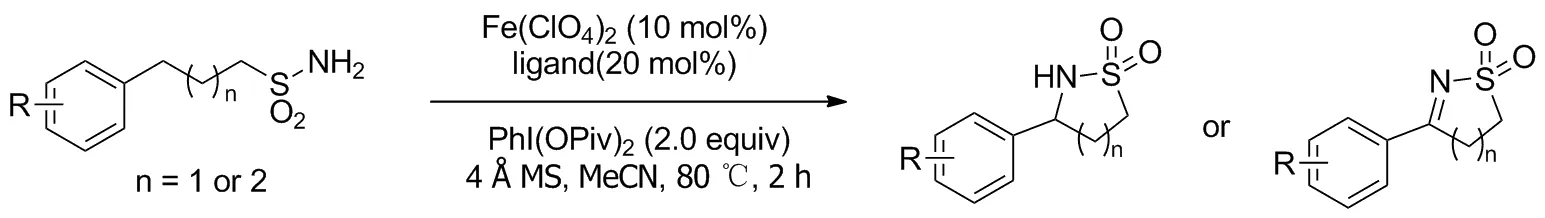
2019年,Zhong等[13]报道了铁催化脂肪族C—H键胺化的例子(Scheme 10),构建药物分子中常见的环磺酰胺骨架。优势在于使用现成的铁催化剂和无需叠氮化的磺酰胺底物。该方案选择性活化一系列苄基型磺酰胺底物硫原子的γ-或δ-位C—H键,产率50%~88%。调控反应条件可以使反应倾向于合成环磺酰胺或环磺酰亚胺,后者产率可达75%~92%。
2019年 ,Pandey等[14]发表了以钌为中心原子的光氧化还原催化苄基位C—H键胺化的方案(Scheme 11),用于合成六元含氮杂环。具有无需导向基团和额外氧化剂,底物无需预处理等优势。可耐受各种氮保护基苄基位的各种取代基,产率38%~84%。推测机理是光照激活光催化剂与底物间发生单电子转移(SET),诱导底物形成自由基,随后发生分子内HAT活化苄基C—H键,最后脱去质子环化得到目标产物,氟试剂selectfluor在反应中作为氧化淬灭剂和氢自由基的受体。
2019年,Bosnidou等[15]报道了碘催化的C—H键胺化的方案(Scheme 12)。以三氟甲磺酰胺为胺化试剂,在光照和单质碘催化下对C(sp3)—H尤其是苄基位进行催化胺化。反应对芳基上各种取代基具有耐受性,产率60%~85%,对药物分子和氨基酸衍生物依然保持良好产率。进一步验证发现通过调控反应条件本方案可以对有两个反应位点的底物实现先分子间再分子内胺化,构建了同一底物经过两次胺化合成含氮杂环的方案。

Scheme 13

Scheme 14
Scheme 15
Scheme 16
Scheme 17
2.3 以胺类化合物为氮源的苄基位C—H键胺化
胺类是最常见易得的氮源,但是胺类氮氢键活泼易发生副反应。所以以胺类作为氮源时通常需要保护基团的参与,如Boc基团、酰基等。亚胺也是一种值得尝试的氮源。2020年,Min等[16]报道了铜催化仲胺选择性远程C(sp3)—H胺化的方案(Scheme 13)。N-氟酰胺底物和二级胺氮源对各种取代基均有耐受性。铜催化剂与胺配位通过SET过程诱导底物N—F键断裂产生自由基中间体,中间体分子内发生1,5-HAT活化远端sp3碳原子。苄基型底物产率为36%~80%,且耐受性、克级反应表现良好。
2020年,Fang等[17]报道了碘介导C(sp3)—H胺化的方案(Scheme 14)。以2′-苄基-2-氨基联苯为底物,碳酸铯和碘单质用于诱导底物产生氮自由基,经历HAT活化苄基碳原子进行环合。环合产物再次与碱和碘作用脱氢芳构化得到菲啶衍生物。底物三个苯环对各种取代基具有耐受性,产率70%~95%。该方案底物易制备,无过渡金属参与,适用于合成广泛的菲啶衍生物。
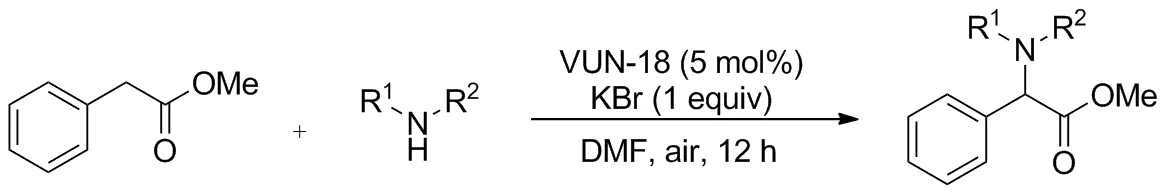
2017年,Tran[18]报道了一种代号VNU-18的五配位铜金属有机骨架,作为一种多相催化剂催化羰基邻位的苄基位C(sp3)—H键胺化,用于合成α-氨基酸酯类化合物(Scheme 15)。该催化剂易于分离,可多次回收利用。底物范围广,当羰基和苄基相连时以47%~73%的产率胺化苄基位C—H键。机理研究表明铜催化剂用于催化底物羰基邻位溴代,随后与氮源偶联得到胺化产物。
Scheme 18
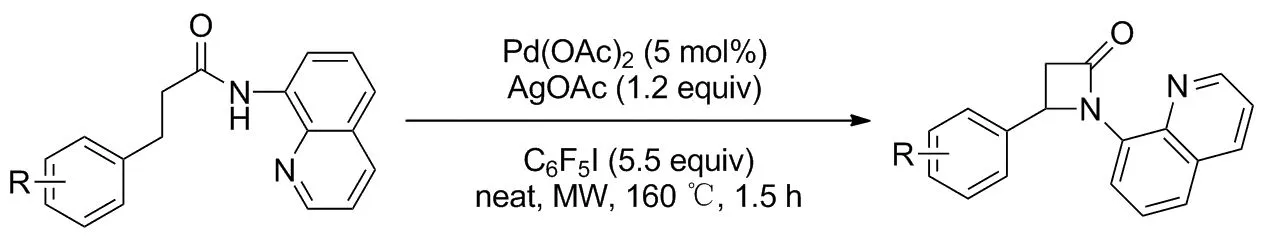
Scheme 19
Scheme 20
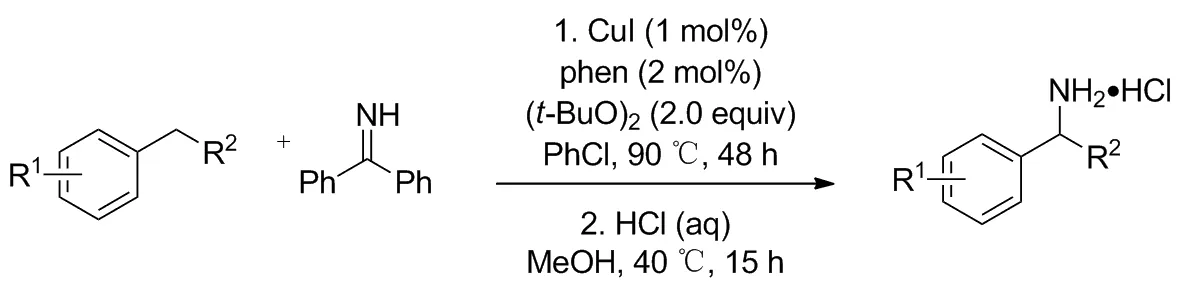
2019年,Kramer[19]发表了一种铜催化合成α-取代苄胺的方法(Scheme 16)。采用交叉脱氢偶联(CDC)反应策略,以常见的二芳基亚胺实现一系列烷基苯苄位C—H键胺化,首次将CDC和二芳基偶联剂结合起来。反应以CuI为催化剂,邻菲罗啉为配体,(t-BuO)2为氧化剂,氯苯为溶剂,第二步加入盐酸溶液及甲醇即可直接的一锅煮得到简单的苄基伯胺盐酸盐,产率38%~67%。 CuI可低至0.1 mol%,成本低廉;反应对空气和水不敏感;无需柱色谱分离。缺点是反应时间过长(48~72 h)。
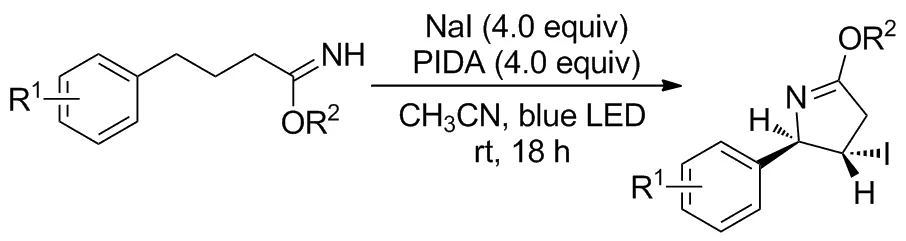
2018年,Kumar等[20]报道了光介导C—H键胺化的范例(Scheme 17)。以4-芳基丁酰亚胺为底物,与活化的碘试剂作用生成碘亚胺,在光照下N—I键裂解形成自由基;经过1,5-HAT活化酰基远端苄基位C—H键,再与碘自由基作用后脱去碘化氢发生环化,最终得到二氢吡咯衍生物。反应可在室温下进行,对不同的烷氧基和芳基取代基有耐受性,当底物含其他反应位点时可以选择性胺化苄基位,产率49%~75%并有良好的非对映选择性(dr值3.2/1~19/1)。另一个意外优势在于使用碘添加剂使产物二氢吡咯C4位均被碘取代,这将为进一步的功能化提供桥梁。
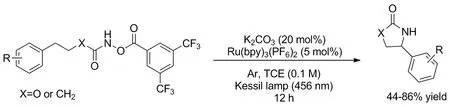
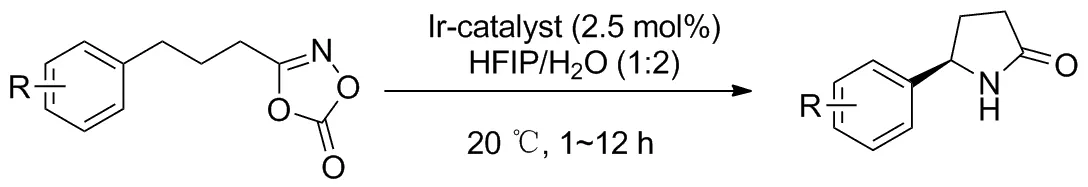
酰胺中的酰基可为胺提供保护,且方便分解,也可作为氮源。2020年,Jung等[21]报道了一个通用性很强的C(sp3)—H催化胺化的方案(Scheme 18),酰胺底物分子内胺化指定位置得到内酰胺,苄基位可顺利地完成。计算和实验筛选出了芳基甲酸酯作为离去基团代替常用的叠氮基团[22],具有方便易得和高效的优势。在光照下完成对苄基位胺化得到一系列噁唑烷酮,产率44%~86%。不含其它杂原子的底物也可顺利完成胺化得到γ-内酰胺,产率45%~84%,这也是钌光催化剂合成γ-内酰胺的第一个例子。
2017年,Zhang等[23]报道了钯催化酰胺β-位C(sp3)—H胺化的方案(Scheme 19)。底物8-氨基喹啉酰胺在醋酸钯催化下β-位C—H键与酰胺氮原子环合得到β-内酰胺衍生物。实验探究得知该方案对β-位为亚甲基的底物非常适用,β-位为苄基位时可耐受芳基上各种取代基,环合产物产率71%~96%。经研究喹啉基团在反应中起了不可替代的作用,而β-位为次甲基或亚甲基附近存在双键和三元、四元环的情况下不能反应。
2.4 其他氮源
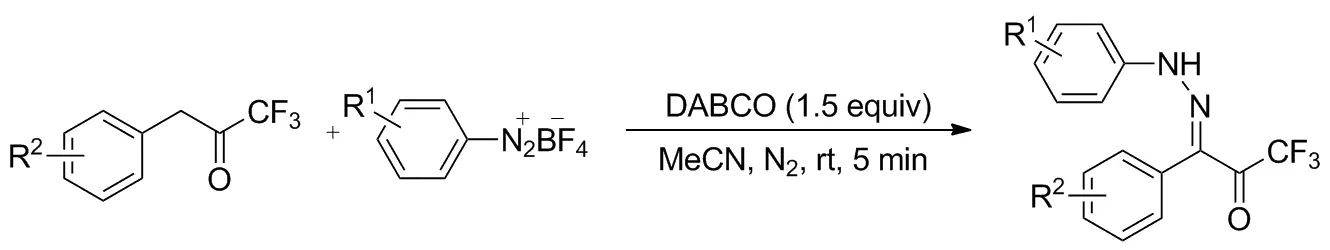
2018年,Zhu等[24]报道了苄基位胺化的方案(Scheme 20)。该方案无需催化剂、导向基团、过渡金属和叠氮化底物,清洁绿色分离方便。反应以苄基三氟甲基酮为底物,四氟硼酸芳基重氮盐既为氮源又做氧化剂,在室温下只需5 min即可以优秀的产率得到苄基位亚胺化产物。底物范围很广,两个芳环上被各种取代基取代均可顺利进行,大部分产率80%~97%,并且E/Z异构体比例均高于20/1。

Scheme 21
Scheme 22

Scheme 23
Scheme 24

Scheme 25
2018年,Clark等[25]报道了锰催化苄基位C—H键胺化的例子(Scheme 21),产率60%左右。该小组创造性地将氮源2,2,2-三氯乙基氨基磺酸酯(TcesNH2)和碘氧化剂PhI(OPiv)2预先反应形成亚胺基碘烷(PhI=NTces),同时充当氮源和氧化剂。除此之外该方案还具有以下特点:反应底物、催化剂和氧化剂均可通过已报道的方法大量制备;底物范围很广且位点电子云密度越高反应活性越强,因此当底物芳基连有强吸电子基团时产率下降;当底物中有多个C(sp3)—H时可以选择性胺化苄基位;更重要的是成功对十多种药物分子骨架进行了胺化,选择性良好,凸显其应用潜力。
2017年,Jeong[26]报道了铑催化喹啉苄基位C—H键胺化的方案(Scheme 22)。以8-甲基喹啉作为底物,偶氮二甲酸酯为氮源,铑催化芳甲基C(sp3)—H键胺化得到目标产物。底物范围广,大部分产率在70%~90%。喹啉在反应中充当导向基团,铑催化剂在与喹啉氮原子和苄基碳原子配位后,再与氮源配位,经历还原消除脱去金属原子胺化苄基位C—H键。
2017年,Li的团队[27]报道了一种统一的钌-光催化剂氧化还原催化苄基和叔碳C(sp3)—H的胺化(Scheme 23)及羟基化的报道。针对反应底物和目标产物不同筛选了各自的最佳条件,但是都使用了超价碘试剂BI-OH(hydroxyl benziodoxole),用于在反应中诱导反应位点形成自由基。自由基与钌催化剂发生SET变为碳正离子,溶剂中的乙腈充当氮源也参与反应,对碳正离子进行亲核捕获得到胺化产物;钌催化剂则在光照下发生氧化还原反应再生。产率62%~74%,条件温和,副产物很少。
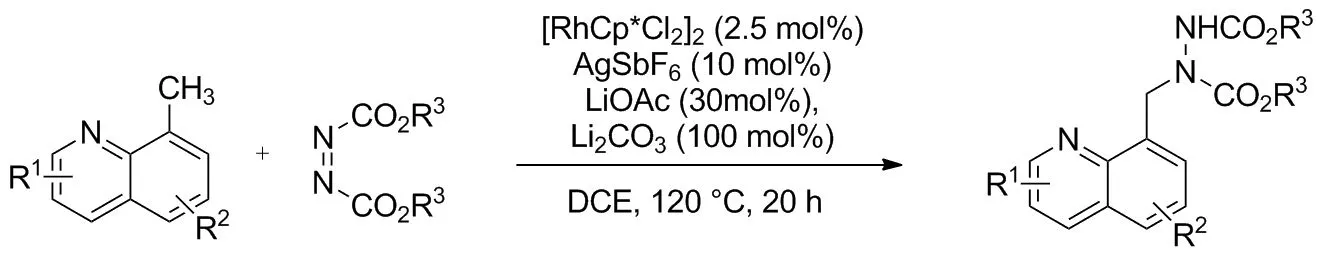
二噁唑酮常在胺化报道中作为氮源,其方便易得,且稳定性好。2019年,Fukagawa等[28]报道了Cp*Rh(III)/手性双萘基羧酸(CCA)联合催化8-乙基喹啉苄基位的对映选择性C(sp3)—H胺化(Scheme 24)。喹啉底物和氮源二噁唑酮也有良好的官能团耐受性,表现出46%~99%的高产率和良好的对映选择性(ee值82%~88%)。喹啉8-位为乙基以外的烷基取代时需要加大添加剂用量和延长反应时间保证产率。
Scheme 26
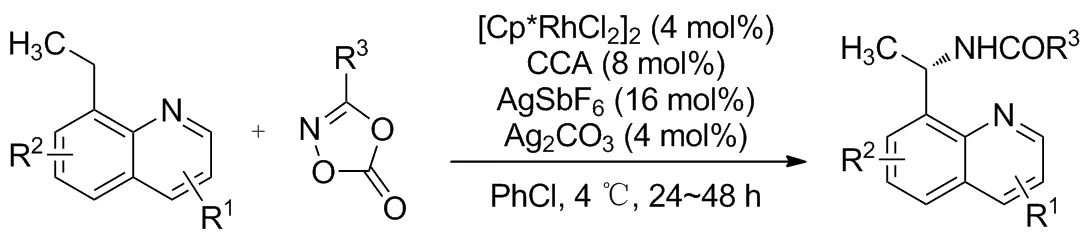
2018年,Hong等[29]发表了一个铱催化C(sp3)—H胺化合成γ-内酰胺的例子(Scheme 25),设计合成并筛选出的铱配合物催化剂经计算和实验验证具有高效的催化活性,制备方便;底物二噁唑酮也可通过羧酸前体方便制得。对苄基位的催化胺化产率53%~99%,芳基的给电子性质更适合反应进行。此外还避免了未加保护的氮原子对催化剂的毒性[30]和金属有机中间体分解为异氰酸酯[31-32]等问题。
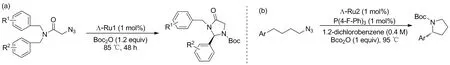
苄基位C—H键胺化很多情况下会涉及到立体化学的问题,通常采用手性催化剂来获得立体选择性。2019年,Zhou等[33]报道了钌催化分子内C—H键胺化的方案。参考此前报道制备出的手性钌配合物催化剂[34](Chart 1),在高温下仍可保持手性,确保高对映选择性。以 2-叠氮-N,N-二苄基乙酰胺为底物,活化苄基位C—H键并以31%~95%的产率提供手性咪唑啉-4-酮(Scheme 26a),这是使用脂肪族叠氮化物的高度对映选择性C(sp3)—H胺化的第一个例子。同年,该课题组[35]报道了对以上钌催化剂的结构调整后与三苯基膦衍生物联合催化的分子内C—H胺化反应(Scheme 26b),以4-叠氮丁苯及其衍生物为底物,合成手性的吡咯烷。弥补了这方面制备产率低选择性低的缺陷[36]。反应历程是先通过膦对叠氮化物的Staudinger反应得到膦亚胺,亚胺转移至钌配合物中心,随后立体选择性地插入苄基位C—H键,得到的钌配位吡咯烷,引入Boc保护基同时脱去钌得到目标产物。本方案对不同取代的底物产率在50%左右,推电子基对反应体系相对友好。尽管产率不高,但产物ee值高达99%,为合成手性芳基吡咯烷酮提供了一种新策略。

Chart 1
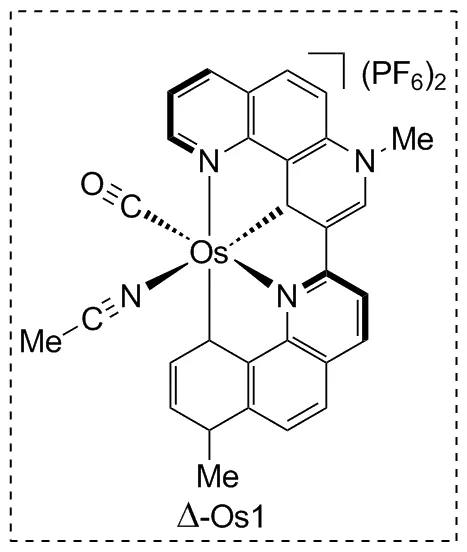
Chart 2
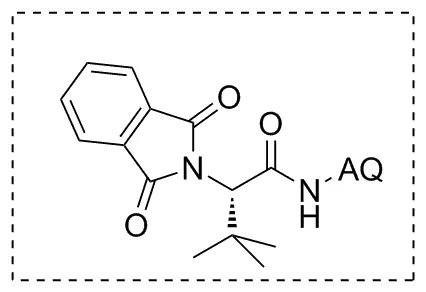
Chart 3
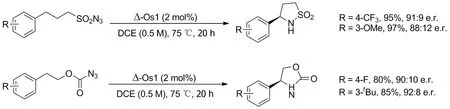
2020年,Wang等[37]报道了一种锇中心原子的手性催化剂(Chart 2),其手性完全来自中心原子而非配体,相比上面的例子该催化剂没有C2对称性。实验验证其可以实现对两种叠氮底物的芳基苄位C(sp3)—H不对称催化胺化(Scheme 27),得到手性氮杂环。对底物的初步拓展中芳基对不同取代基具有耐受性,产率80%~99%,对映选择性优秀,优于该小组最近开发的结构相似的钌中心原子手性催化剂。
Scheme 27

Scheme 28
Scheme 29
以上两个例子所用的催化剂手性来源于中心原子,通过设计手性配体也可实现不对称催化胺化。2017年,Munnuri等[38]报道了铑催化C(sp3)—H胺化的例子(Scheme 28),用于构建取代手性吡咯烷。其特点在于产物空间结构可调控,设计筛选了两种铑催化剂的位阻起到了决定性作用。以苄基型底物为例,催化剂的配体体积大小决定了对底物芳基的排斥作用的强弱,导致氮原子与铑配位形成的氮烯插入不同的苄基位C—H键,最终导致合成顺式和反式两种2,5-二取代吡咯烷,产率中等(46%~62%),有良好的非对映选择性。反应底物无需导向基团,产物氮原子无保护基,并且室温下即可反应。
依靠位阻的空间排斥作用是确保对映选择性的有力手段,除此之外还有很多比较弱的非共价相互作用如氢键、范德华力、疏水作用等,利用这些相互作用来构建手性化合物的报道还很少。2019年,Wang等[39]报道了一个通用性铱催化C(sp3)—H胺化的例子(Scheme 29),创造性地利用了多种非共价相互作用机制,提供了第一条在温和条件下不对称合成手性γ-烷基/芳基-γ-内酰胺的通用路线。该方案中配体(Chart 3)的氨基喹啉基(AQ)和邻苯二甲酰基在铱中心原子周围形成手性疏水口袋,允许反应能在水或极性溶剂进行。产物的对映选择性则依靠底物进入口袋后受到的多种作用。如苄基型底物的苯基与AQ之间存在π-π堆积作用,脂肪族底物则存在C—H键与π电子云的相互作用;底物与邻苯二甲酰基的羰基之间存在范德华力的作用等。该方案苄基位C—H键的胺化表现优秀,大部分产率在80%~95%。对连有强吸电子基的或邻位取代的芳基底物产率中等,同时有着很好的对映选择性(ee值86%~99%)。
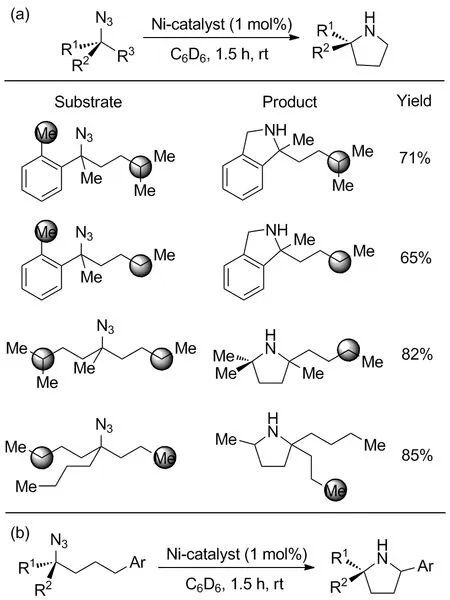
不同反应位点的反应活性区别很大,原因在于不同的碳原子周围的电子云密度不同,C—H键的解离能不同,因此反应中间体的稳定性也不同。本文讨论的苄基位是反应活性较高的位点,一般在多个位点共存时会优先发生反应。例如2020年,Dong等[40]报道的镍催化C—H键胺化的方案,用于构建饱和含氮杂环。设计了镍配合物催化剂用于激活叠氮基团产生氮自由基,通过氢原子提取(HAA)活化叠氮基δ-位C(sp3)—H键,最终与氮原子环合得到吡咯烷。实验验证反应的区位选择性与各位点C—H键解离能相符。在多个反应位点共存时,解离能小的C—H键被选择性活化,解离能大的C—H键位点未被检测到发生反应(Scheme 30a)。苄基型底物反应顺利,在温和条件下2 h内得到目标产物,产率65%~95%(Scheme 30b)。
Scheme 30
Scheme 31
克服底物的先天反应倾向是C—H键活化领域面临的挑战。在不同空间或电子环境中,C—H键之间的正交位置选择性可以通过改变过渡金属来实现。Fiori小组研究发现铑催化剂Rh2Ln倾向于在三级碳发生胺化[41];而Paradine的小组设计的锰催化剂倾向于胺化苄基位[42]。除此之外通过筛选催化剂的配体,利用催化剂空间性质改变选择性也是一种重要策略。反应活性较强的苄基和三级碳C—H键依赖于周围位阻较大的芳基或多个甲基亚甲基的共轭效应,反过来利用位阻能够实现选择性活化活性较低的一级、二级碳。2019年,Bakhoda等[43]报道了合成一种大位阻铜配合物催化剂用于催化C(sp3)—H胺化(Scheme 31a),特点是底物中多个反应位点共存时选择性胺化更难反应的一级二级碳而非富电子的三级碳。底物探究验证了这一选择性,以中等至良好的产率得到带保护基团的胺化产物。其中几种苄基型底物产率为57%~78%(Scheme 31b)。
2017年,Corbin等[44]发表了一篇银催化叔碳C—H键胺化的报道(Scheme 32),探究当底物中含有多个相似的叔碳反应位点时,不同银配合物催化剂对不同位点的催化倾向。在合成的一系列银配合物催化剂中筛选出了可以选择性催化活化苄基位叔碳、异丙基叔碳等不同叔C—H键的催化剂。其中一种“五吡啶二甲基”的配体与银的配合物催化剂可以高效地选择性催化活化苄基位叔C—H键,产率80%~90%。目前金属原子和配体之间微妙的电子、位阻因素对反应位点的影响关系仍不明确,但是这种调控反应位点的方式是可行的,并且具有应用价值。
综上,从氮源、立体选择性和区域选择性3个角度筛选介绍了近5年有关苄基位C—H键胺化的报道,虽然很多并非仅适用于苄基位,但也引用了其中涉及苄基位胺化的部分进行了介绍,并突出了报道的特色。总体来说苄基位C—H键胺基化反应理念先进、产物应用广泛,且得益于苄基位自身的高反应活性得到大量关注,相关报道数目众多。其中常用策略主要包括以下几种:一是过渡金属催化,通常以过渡金属配合物作为催化剂进行催化。过渡金属可在反应中插入苄基位C—H键后经历氧化加成-还原消除过程完成胺化,并且配体还会影响产物的选择性;二是无过渡金属催化。碘及其衍生物是常被用到的催化剂或反应剂,多数通过自由基机理完成胺化;此外还有综合性较强的过渡金属光催化剂催化、金属自由基催化等策略。不同策略优劣各异。过渡金属催化产率高,还能对选择性进行调控,但使用过渡金属会对环境造成较大污染,催化剂的合成和表征也比较繁琐;无过渡金属催化策略清洁绿色,后处理更加方便,但产率偏低,此外对产物手性有要求的方案中这种策略运用很少。运用这些策略的苄基位C—H键胺化方案很多在合成领域已有良好的表现,体现出这一方向的应用潜力。但同时过渡金属催化的污染问题,无过渡金属催化的产率和选择性问题,两者的取长补短等仍是目前苄基位胺化存在的问题,还有很大的发展空间等待进一步研究。

Scheme 32
不仅限于苄基位胺化,C—H键活化拥有高原子经济性,符合绿色化学的发展理念,有蓬勃的发展活力。人们正在从产率、选择性、成本、环境等多方面建立或优化这方面的反应体系,并在医药、材料、农业等方面得到应用,从而提高生产效率。
