Bethlem 肌病一家系临床表型及基因突变分析
2021-04-27张琼哲吴世陶石伟伟刘恒方
张琼哲 吴世陶 崔 明 张 敏 石伟伟 刘恒方
郑州大学第五附属医院,河南 郑州 450052
Bethlem肌病是先天性肌营养不良的一种亚型,是由细胞外基质中Ⅵ型胶原蛋白基因的突变导致,多数病例呈常染色体显性遗传,可于较小年纪发病,主要表现有四肢近端逐渐出现的肌肉无力、抬腿或抬起手臂、蹲起动作困难,血清肌酸激酶水平升高及手指、腕、肘、踝关节的挛缩,肌电图呈肌源性损害[1-3]。目前该病在我国比较少见,无法统计出发病率[4-6]。本文报告郑州大学第五附属医院收治的一Bethlem肌病家系的临床表现及相关检查检验结果,通过查阅相关文献,分析总结该病的特点,以增强医务人员对该病的认识及鉴别。
1 资料与方法
1.1 临床资料 患者1(Ⅲ12),即先证者,女性,2001年出生,于2017-03-30以“进行性四肢无力3 a”为主诉首次于郑州大学第五附属医院神经内科门诊就诊。患者3 a前无明显诱因出现双上肢抬起无力,无法同时抬起,几乎同时发现右下肢腿径变细,后逐渐出现双下肢无力,右下肢无力更为明显,表现为在学校跑操时频繁摔倒,跑操15 min 平均摔倒3~4次,上楼梯和蹲起困难,就诊时已无法进行体育活动,无法跑步,但仍能行走,无感觉异常,否认夜间呼吸困难等呼吸肌受累表现。于当地医院行肌电图检查提示肌源性损害,为明确诊断来我院就诊。患者系足月顺产第一胎,出生时体质量为正常范围,产伤史、窒息史均无,1岁2个月可独立行走,1岁6个月会说话,幼年期各项功能与同龄者无明显差异,在校学习中等。母亲与其症状相似但较先证者更重,父亲身体健康,否认有近亲婚史。门诊专科体格检查:智力正常,双侧鼓腮无力,余脑神经检查未见异常,双上肢及右下肢近端肌力4 级,左下肢近端肌力5-级,四肢远端肌力5 级;四肢肌张力均降低,腱反射未引出,双侧掌颌反射及巴宾斯基征阴性;双侧深浅感觉正常;右下肢大腿肌肉萎缩,腿径变细,以臀大肌、股直肌、股外侧肌、股内侧肌、股二头肌、耻骨肌、内收肌为主;双侧翼装肩胛,双足跟腱挛缩,可足尖行走,足跟行走欠佳,Gowers 征阳性;双侧指鼻试验、跟膝胫试验阴性;颈软,无抵抗,脑膜刺激征阴性。
实验室检查(2020-07-15):血尿粪常规、肝肾功能、电解质、凝血功能正常,乳酸脱氢酶(LDH)257 U/L(114~240 U/L),α-羟丁酸脱氢酶(HBDH)187 U/L(72~182 U/L),肌酸激酶(CK)485 U/L(24~183 U/L),肌酸激酶同工酶(CK-MB)70.0 U/L(0~25.0 U/L)。心脏彩超未见明显异常,心电图示正常窦性心律。
患者2(先证者母亲,Ⅱ7),1982 年出生,与先证者首次于2017-03-30 于郑州大学第五附属医院就诊,就诊时35 岁,自诉11 岁时发现右上肢变细、无力,14 岁时逐渐出现双下肢无力、行走异常,17 岁时逐渐出现行走姿势异常,表现为躯体前弓,行走不稳,2017 年摔伤后不能行走,后一直坐轮椅,平素借助工具可自行穿衣、生活,现因髋关节、膝关节挛缩明显无法伸直平躺于床上,丧失行走能力,胸腰椎侧弯明显,双侧翼装肩胛,Gowers征阳性。除生育先证者外,还有一次生育史,为先证者妹妹(图1),有与先证者类似的肌无力和关节活动障碍症状。该患者父母身体健康,否认近亲婚配,其一兄一姐均体健。门诊专科体格检查:高级智能正常,鼓腮无力,髋伸、屈3级,膝伸、屈4级(图2),患者双下肢近端肌力3级,远端肌力5 级,左下肢近端肌力5-级,右下肢近端肌力4级,双下肢远端肌力5级,肌张力降低,四肢腱反射未引出,双侧病理征阴性,双侧深浅感觉正常,双侧指鼻试验、跟膝胫试验无法查。颈软,无抵抗,脑膜刺激征阴性。
实验室检查(2020-07-15):血尿粪常规、肝肾功能、电解质、凝血功能正常,LDH 176 U/L(114~240 U/L),HBDH 131 U/L(72~182 U/L),CK 179 U/L(24~183 U/L),CK-MB 24.2 U/L(0~25.0 U/L)。心脏彩超未见明显异常,心电图示正常窦性心律。
患者3(先证者妹妹,Ⅲ13),女性,2004 年出生,本次因“右侧肢体进行性无力6 a,行走姿势异常1 a”于2020-07-15 再次与先证者及其母亲于我院就诊。患者10 岁左右时擦黑板时发现右上肢抬起无力,12 岁时同学发现其跑步姿势异常,未在意,15 岁跑操时发现跟不上跑步队伍,且右下肢较左下肢短,行走时身体不平衡,跑步时更明显,自行身体锻炼较前有所好转,仍可继续上操跑步,患者足月顺产第二胎,出生时体质量正常,无产伤和窒息史,1岁时可独立行走,1 岁4 个月会说话,幼年期各项机能与同龄者无明显差异,学习中上等。门诊专科体格检查:高级智能正常,双侧鼓腮无力,余脑神经检查未见异常,右侧上下肢近端肌力5-级,远端肌力5级,左侧肢体肌力5级;四肢肌张力均降低,腱反射未引出,双侧掌颌反射及巴宾斯基征阴性;双侧深浅感觉正常;左侧腓肠肌肥大,无肌肉压痛;双侧翼装肩胛,双足跟腱挛缩,可足尖行走,足跟行走欠佳,Gowers征阴性;双侧指鼻试验、跟膝胫试验阴性;颈软,无抵抗,脑膜刺激征阴性。
实验室检查(2020-07-15):血尿粪常规、肝肾功能、电解质、凝血功能正常,谷草转氨酶43 U/L(8~40 U/L),LDH 357 U/L(114~40 U/L),HBDH 228 U/L(72~182 U/L),CK 936 U/L(24~183 U/L),CK-MB 94.2 U/L(0~25.0 U/L)。心脏彩超未见明显异常,心电图示正常窦性心律。
1.2 肌肉活检 先证者股直肌活检肌肉病理:HE染色示可见肌纤维大小不一,部分肌纤维圆形化,较多变性、坏死肌纤维,少量不透光肌纤维,结缔组织增生(图3)。
1.3 基因检测 采集受检者外周血3 mL,提取基因组DNA,应用探针杂交捕获神经肌肉病相关基因序列[7](北京信诺佰世医学检验所),通过下代高通量测序仪(Illumina)进行测序。其测序后的数据运用NextGene V2.3.4 软件与UCSC 数据库提供的人类基因组hg19 参考序列进行过滤、比对等分析处理。采用Sanger测序对明确或可能相关的基因变异进行验证。先证者之妹对其目标基因进行验证(北京金准基因科技有限公司)。见图4。
2 讨论
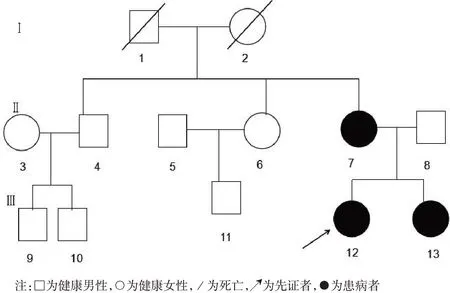
图1 家系图Figure 1 Family diagram
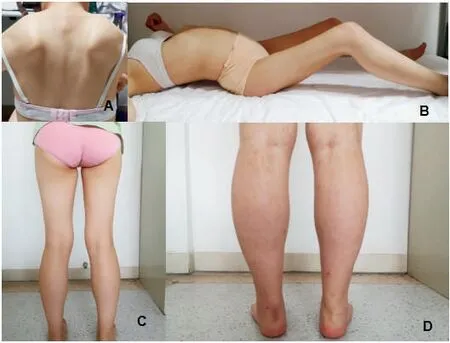
图2 A:先证者母亲,翼装肩胛;B:先证者母亲,腰椎前凸,髋、膝关节挛缩;C:先证者,右下肢腿径变细;D:先证者妹妹,右下肢肌肉萎缩Figure 2 A is the protester’s mother, wing-mounted shoulder blade; B is the mother of the protestor, with lumbar lordosis and contracture of hip and knee; C is the protestor, the leg diameter of the right lower limb becomes thinner; D is the sister of the protester. Muscle atrophy of the right lower limb
Ⅵ型胶原蛋白相关肌病是由于Ⅵ型胶原蛋白功能缺陷导致的遗传性肌病,有肌肉无力萎缩、关节挛缩的特点。大致分为四类临床表型:Bethlem 肌病(Bethlem myopathy,BM,MIM158810)、Ullrich 先天性肌营养不良[8](Ullrich congenital muscular dystrophy,UCMD,MIM254090)、肢带型肌营养不良症和遗传性硬化性肌病,后两者与BM 类似,症状介于前两者之间,发病数极少,归为同一谱系疾病。Ⅵ型胶原蛋白属于细胞外基质蛋白,广泛存在于肌肉、肌腱、皮肤、软骨和椎间盘等组织的细胞外基质中,从而形成与基底膜密切相关的微纤维网络[9-11]。有活性的Ⅵ型胶原蛋白是在分别由COL6A1(NM_001848)、COL6A2(NM_ 001849)和COL6A3(NM_004369)基因编码的α1、α2、α33条不同的肽链基础上形成,通过三螺旋结构形成单体,然后再形成四聚体[12-13]。无论哪个基因发生致病性变异,该蛋白功能就会出现异常,从而引发某种相关肌病。
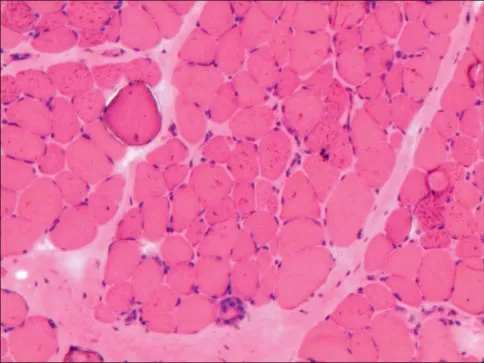
图3 伊红-苏木素染色示结缔组织增生、脂肪组织取代肌组织Figure 3 Eosin-hematoxylin staining shows connective tissue hyperplasia with adipose tissue replacing muscle tissue
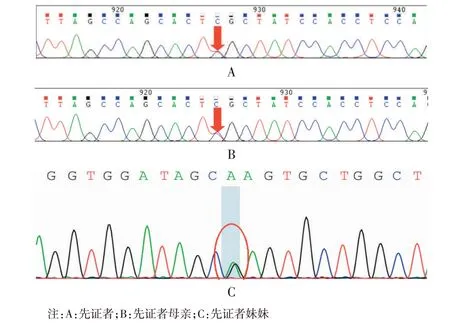
图4 先证者及其母、其妹存在相同基因突变,即COL6A3基因错义突变c.4270C>TFigure 4 The proband, his mother and his sister had the same gene mutation, namely COL6A3 missense mutation C. 4270C>T
在这4类亚型中,UCMD表型最重[14]。患者于婴幼儿期即可出现轴性和近端关节挛缩、肌无力,以及远端关节过松,症状逐渐加重,后期多不能行走,多需呼吸机辅助通气。相对而言Bethlem 肌病表型较轻,其遗传方式多以常染色体显性为主,也有纯合或复合杂合突变致病[15-17]。多于年纪较小时逐渐出现近端肌无力或关节畸形,缓慢进展。与前者不同的是,Bethlem肌病患者呼吸肌基本不受累。据文献[18-19]报道总结出Bethlem肌病诊断标准:(1)家族史,常染色体显性或隐性遗传;(2)出生后可能出现肌张力低、远端关节松弛、髋关节脱位、关节挛缩或脊柱侧弯等;(3)发病年龄,多于20岁前发病,也有成年后逐渐出现症状者;(4)影响四肢近端肌肉力量及肌肉挛缩,多以近端肌肉发病,逐渐加重,缓慢进展,肩胛带肌及下肢肌萎缩,最终可广泛肌无力[20-21];(5)结缔组织受累,可有不同程度的关节松弛、挛缩;(6)中枢神经系统或心脏、呼吸功能一般不受累;(7)血清学肌酶通常轻度升高,也可正常或10 倍以上升高,肌电图可见MUAP 窄小,肌肉病理可见非特异性肌源 性 损 害;(8)基 因 检 测 示COL6A1、COL6A2、COL6A3突变[22-24]。
本文报道一Bethlem 肌病家系,3 例患者均存在COL6A3基因c.4270C>T突变,该样本在此基因外显子区域发现一处杂合突变:c.4270C>T(胞嘧啶>胸腺嘧啶),导致氨基酸改变p.R1424C(精氨酸>半胱氨酸)。HGMDpro 数据库报道情况:变异位点c.4270C>T 未见报道。本研究先证者COL6A3 基因存在杂合变异,其母、妹均存在相同的杂合变异位点,其父情况未知。根据ACMG评级变异指南,该变异评级为可能致病性变异。该样本遵循常染色体显性遗传规律,先证者之父未行基因检测,但根据先证者及其母和妹的基因检测结果,并不影响遗传信息学分析。先证者外婆及大姨、舅舅均无临床症状,考虑先证者母亲为基因突变,并将突变基因遗传给两个女儿,该家系中先证者母亲表型最重,幼年即出现右上肢及双下肢无力,后逐渐出现髋、膝关节的挛缩,脊柱侧弯,现已无法行走。先证者症状较母亲稍轻,先证者妹妹症状最轻,但后两者病情仍处于活动期,逐渐进展,仍需进一步随访。据家属最新回馈结果,先证者妹妹现已无法上操跑步,也印证了基因遗传病其临床表现在家族中有一定异质性,尽管为同一个基因突变所致蛋白功能异常,其引起的临床症状和病情严重程度也可能不同,甚至可无明显临床症状[25-26]。
目前对Bethlem肌病上尚无明确有效方法,由于肌营养不良亚型较多[27-28],症状复杂多样,单从症状区分诊断难度较大,通过基因测序可以一次性检测多个基因突变,提高了诊断精准度,从而诊断出Bethlem肌病也越来越多,由于该病是一种遗传性疾病,早期发现、早期诊断可减少患者不必要的经济负担,同时可扩展该病的基因谱,有助于产前诊断,必要时可终止妊娠减少遗传病的发生率。
